31
Porphyrins & Bile Pigments
OBJECTIVES
After studying this chapter, you should be able to:
![]() Know the relationship between porphyrins and heme
Know the relationship between porphyrins and heme
![]() Be familiar with how heme is synthesized
Be familiar with how heme is synthesized
![]() Understand the causes and general clinical pictures of the various porphyrias
Understand the causes and general clinical pictures of the various porphyrias
![]() Know how bilirubin is derived from heme and how it is handled in the body
Know how bilirubin is derived from heme and how it is handled in the body
![]() Understand the nature of jaundice and appreciate how to approach determining its cause in a patient.
Understand the nature of jaundice and appreciate how to approach determining its cause in a patient.
BIOMEDICAL IMPORTANCE
The biochemistry of the porphyrins and of the bile pigments is presented in this chapter. These topics are closely related, because heme is synthesized from porphyrins and iron, and the products of degradation of heme are the bile pigments and iron.
Knowledge of the biochemistry of the porphyrins and of heme is basic to understanding the varied functions of hemoproteins (see below) in the body. The porphyrias are a group of diseases caused by abnormalities in the pathway of biosynthesis of the various porphyrins. Although porphyrias are not very prevalent, physicians must be aware of them. A much more prevalent clinical condition is jaundice, due to elevation of bilirubin in the plasma. This elevation is due to overproduction of bilirubin or to failure of its excretion and is seen in numerous diseases ranging from hemolytic anemias to viral hepatitis and to cancer of the pancreas.
METALLOPORPHYRINS & HEMOPROTEINS ARE IMPORTANT IN NATURE
Porphyrins are cyclic compounds formed by the linkage of four pyrrole rings through methyne (=HC—) bridges (Figure 31–1). A characteristic property of porphyrins is the formation of complexes with metal ions bound to the nitrogen atom of the pyrrole rings. Examples are the iron porphyrins such as heme of hemoglobin and the magnesium-containing porphyrin chlorophyll, the photosynthetic pigment of plants.
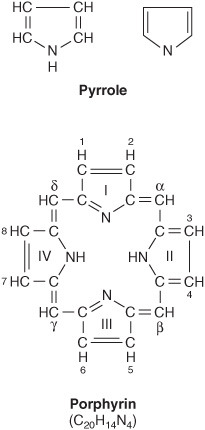
FIGURE 31–1 The porphyrin molecule. Rings are labeled I, II, III, and IV. Substituent positions on the rings are labeled 1, 2, 3, 4, 5, 6, 7, and 8. The methyne bridges (=HC—) are labeled α, β, γ, and δ. The numbering system used is that of Hans Fischer.
Proteins that contain heme (hemoproteins) are widely distributed in nature. Examples of their importance in humans and animals are listed in Table 31-1.
TABLE 31–1 Examples of Some Important Human and Animal Hemoproteins1
Natural Porphyrins Have Substituent Side Chains on the Porphyrin Nucleus
The porphyrins found in nature are compounds in which various side chains are substituted for the eight hydrogen atoms numbered in the porphyrin nucleus shown in Figure 31–1. As a simple means of showing these substitutions, Fischer proposed a shorthand formula in which the methyne bridges are omitted and each pyrrole ring is shown as in Figure 31–2, with the eight substituent positions numbered as indicated. Various porphyrins are represented in Figures 31-2, 31-3, and 31-4.
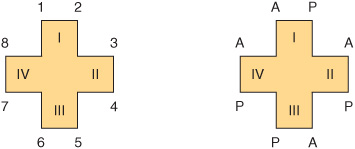
FIGURE 31–2 Uroporphyrin III. (A [acetate]=—CH2COOH; P [propionate] =—CH2CH2COOH.) Note the asymmetry of substituents in ring IV (see the text).
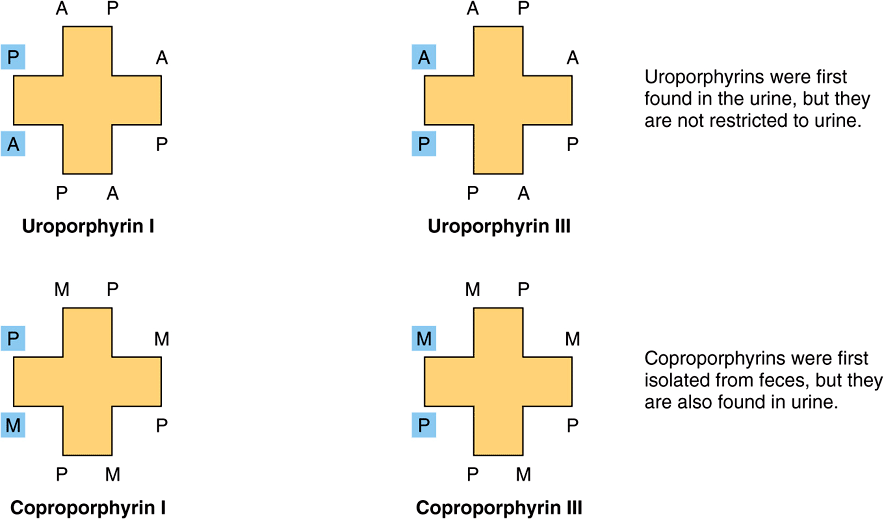
FIGURE 31–3 Uroporphyrins and coproporphyrins. (A, acetate; M, methyl; P, propionate.)
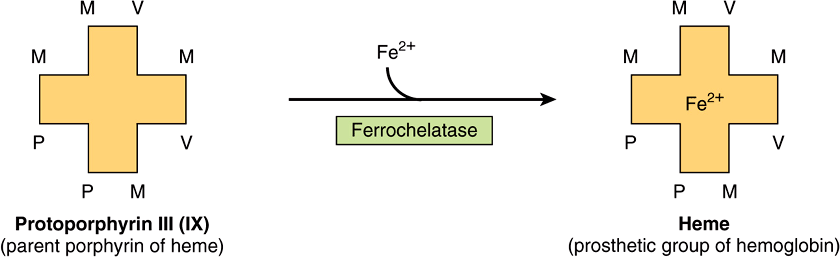
FIGURE 31–4 Addition of iron to protoporphyrin to form heme. ![]()
The arrangement of the acetate (A) and propionate (P) substituents in the uroporphyrin shown in Figure 31–2 is asymmetric (in ring IV, the expected order of the A and P substituents is reversed). A porphyrin with this type of asymmetric substitution is classified as a type III porphyrin. A porphyrin with a completely symmetric arrangement of the substituents is classified as a type I porphyrin. Only types I and III are found in nature, and the type III series is far more abundant (Figure 31–3) and more important because it includes heme.
Heme and its immediate precursor, protoporphyrin IX (Figure 31–4), are both type III porphyrins (ie, the methyl groups are asymmetrically distributed, as in type III coproporphyrin). However, they are sometimes identified as belonging to series IX, because they were designated ninth in a series of isomers postulated by Hans Fischer, the pioneer worker in the field of porphyrin chemistry.
HEME IS SYNTHESIZED FROM SUCCINYL–CoA & GLYCINE
Heme is synthesized in living cells by a pathway that has been much studied. The two starting materials are succinyl-CoA, derived from the citric acid cycle in mitochondria, and the amino acid glycine. Pyridoxal phosphate is also necessary in this reaction to “activate” glycine. The product of the condensation reaction between succinyl-CoA and glycine is α-amino-β-ketoadipic acid, which is rapidly decarboxylated to form δ-aminolevulinate (ALA) (Figure 31–5). This reaction sequence is catalyzed by ALA synthase, the ratecontrolling enzyme in porphyrin biosynthesis in the mammalian liver. Synthesis of ALA occurs in mitochondria. In the cytosol, two molecules of ALA are condensed by the enzyme ALA dehydratase to form two molecules of water and one of porphobilinogen (PBG) (Figure 31–5). ALA dehydratase is a zinc-containing enzyme and is sensitive to inhibition by lead, as can occur in lead poisoning.
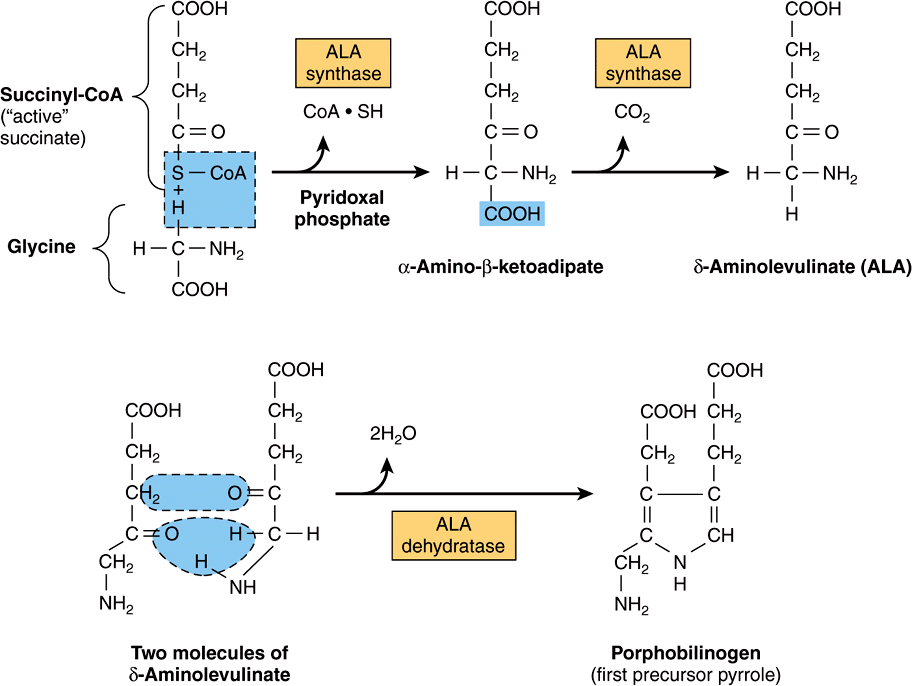
FIGURE 31–5 Biosynthesis of porphobilinogen. ALA synthase occurs in the mitochondria, whereas ALA dehydratase is present in the cytosol.
The formation of a cyclic tetrapyrrole—ie, a porphyrin—occurs by condensation of four molecules of PBG (Figure 31–6). These four molecules condense in a head-to-tail manner to form a linear tetrapyrrole, hydroxymethylbilane (HMB). The reaction is catalyzed by uroporphyrinogen I synthase, also named PBG deaminase or HMB synthase. HMB cyclizes spontaneously to form uroporphyrinogen I (left-hand side of Figure 31–6) or is converted to uroporphyrinogen III by the action of uroporphyrinogen III synthase (right-hand side of Figure 31–6). Under normal conditions, the uroporphyrinogen formed is almost exclusively the III isomer, but in certain of the porphyrias (discussed below), the type-I isomers of porphyrinogens are formed in excess.
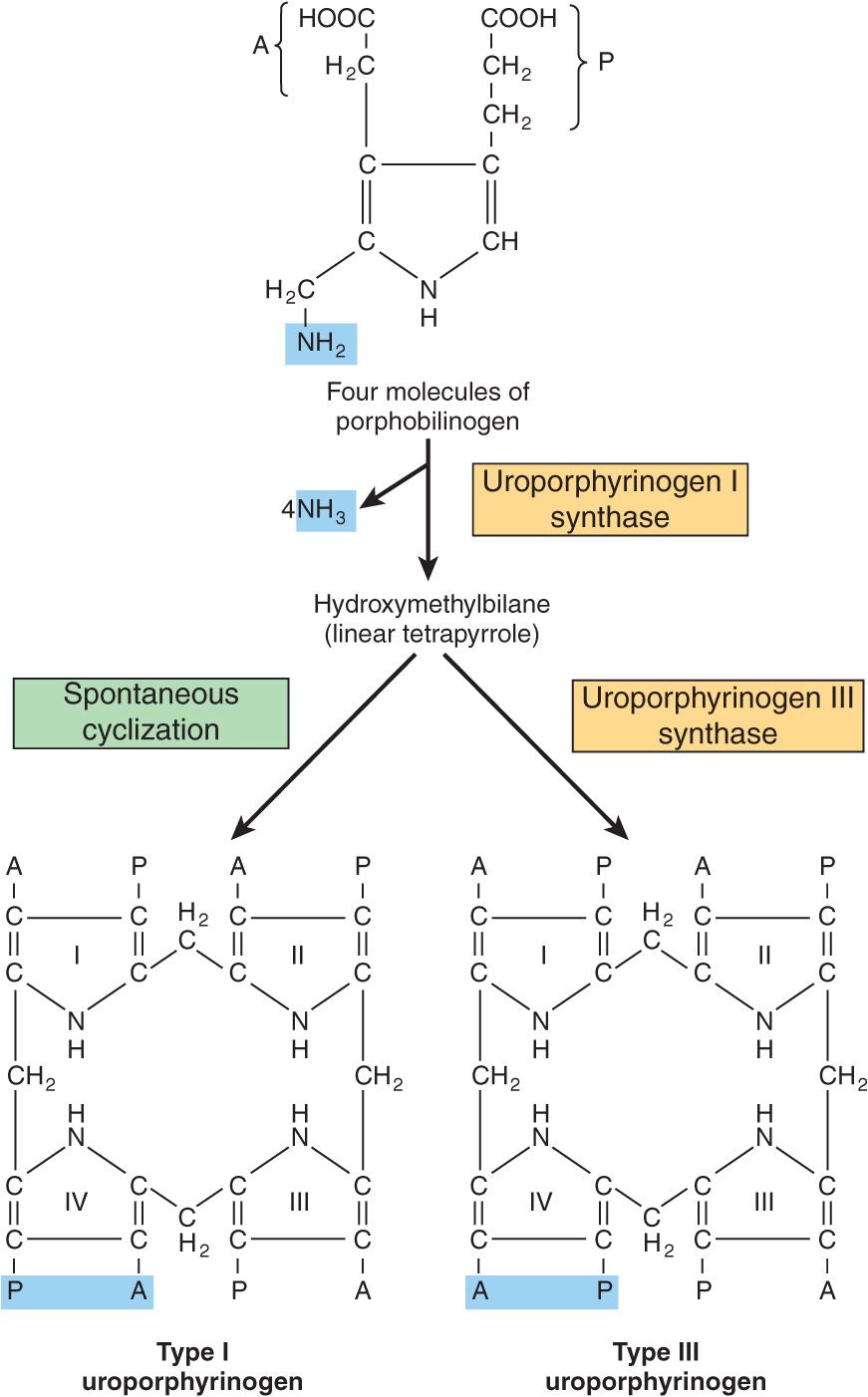
FIGURE 31–6 Conversion of porphobilinogen to uroporphyrinogens. Uroporphyrinogen synthase I is also called porphobilinogen (PBG) deaminase or hydroxymethylbilane (HMB) synthase.
Note that both of these uroporphyrinogens have the pyrrole rings connected by methylene bridges (—CH2—), which do not form a conjugated ring system. Thus, these compounds are colorless (as are all porphyrinogens). However, the porphyrinogens are readily auto-oxidized to their respective colored porphyrins. These oxidations are catalyzed by light and by the porphyrins that are formed.
Uroporphyrinogen III is converted to coproporphyrinogen III by decarboxylation of all of the acetate (A) groups, which changes them to methyl (M) substituents. The reaction is catalyzed by uroporphyrinogen decarboxylase, which is also capable of converting uroporphyrinogen I to coproporphyrinogen I (Figure 31–7). Coproporphyrinogen III then enters the mitochondria, where it is converted to protoporphyrinogen III and then to protoporphyrin III. Several steps are involved in this conversion. The mitochondrial enzyme coproporphyrinogen oxidase catalyzes the decarboxylation and oxidation of two propionic side chains to form protoporphyrinogen. This enzyme is able to act only on type-III coproporphyrinogen, which would explain why type I protoporphyrins do not generally occur in nature. The oxidation of protoporphyrinogen to protoporphyrin is catalyzed by another mitochondrial enzyme, protoporphyrinogen oxidase. In the mammalian liver, the conversion of coproporphyrinogen to protoporphyrin requires molecular oxygen.
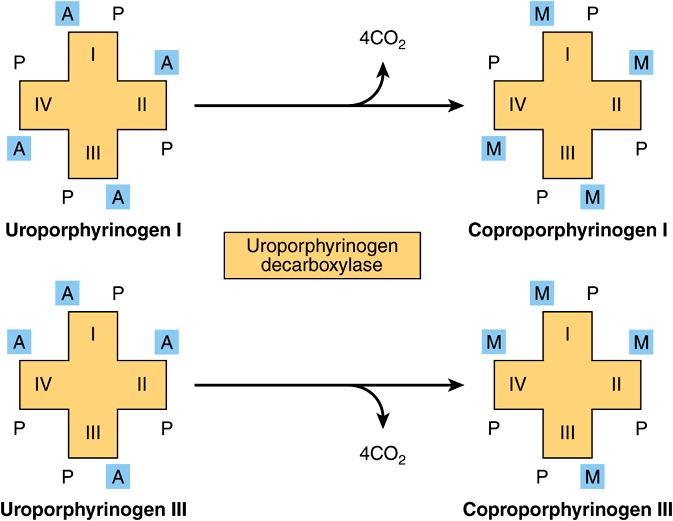
FIGURE 31–7 Decarboxylation of uroporphyrinogens to coproporphyrinogens in cytosol. (A, acetyl; M, methyl; P, propionyl.)
Formation of Heme Involves Incorporation of Iron into Protoporphyrin
The final step in heme synthesis involves the incorporation of ferrous iron into protoporphyrin in a reaction catalyzed by ferrochelatase (heme synthase), another mitochondrial enzyme (Figure 31–4).
A summary of the steps in the biosynthesis of the porphyrin derivatives from PBG is given in Figure 31–8. The last three enzymes in the pathway and ALA synthase are located in the mitochondrion, whereas the other enzymes are cytosolic. Both erythroid and nonerythroid (“housekeeping”) forms of ALA synthase are found. Heme biosynthesis occurs in most mammalian cells with the exception of mature erythrocytes, which do not contain mitochondria. However, approximately 85% of heme synthesis occurs in erythroid precursor cells in the bone marrow and the majority of the remainder in hepatocytes.
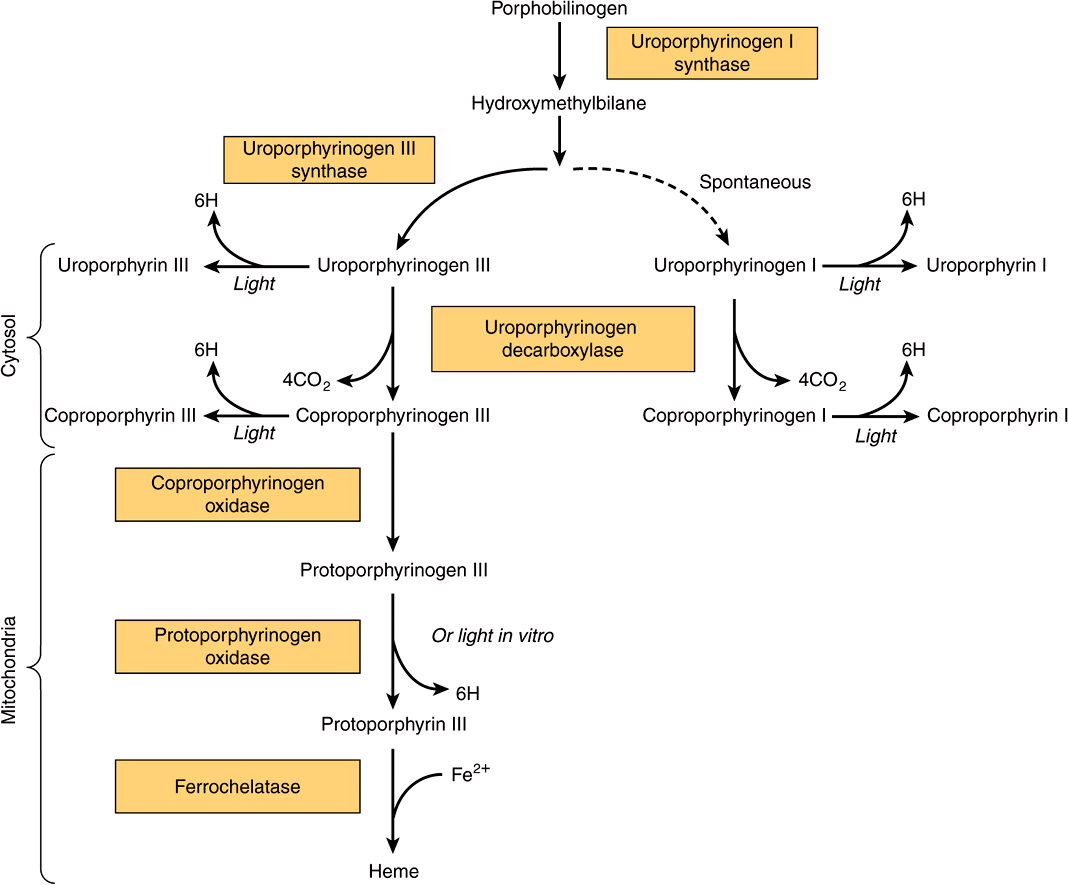
FIGURE 31–8 Steps in the biosynthesis of the porphyrin derivatives from porphobilinogen. Uroporphyrinogen I synthase is also called porphobilinogen deaminase or hydroxymethylbilane synthase.
The porphyrinogens described earlier are colorless, containing six extra hydrogen atoms as compared to the corresponding colored porphyrins. These reduced porphyrins (the porphyrinogens) and not the corresponding porphyrins are the actual intermediates in the biosynthesis of protoporphyrin and of heme.
ALA Synthase Is the Key Regulatory Enzyme in Hepatic Biosynthesis of Heme
ALA synthase occurs in both hepatic (ALAS1) and erythroid (ALAS2) forms. The rate-limiting reaction in the synthesis of heme in liver is that catalyzed by ALAS1 (Figure 31–5), a regulatory enzyme. It appears that heme, probably acting through an aporepressor molecule, acts as a negative regulator of the synthesis of ALAS1. This repression-derepression mechanism is depicted diagrammatically in Figure 31–9. Thus, the rate of synthesis of ALAS1 increases greatly in the absence of heme and is diminished in its presence. The turnover rate of ALAS1 in the rat liver is normally rapid (half-life about 1 h), a common feature of an enzyme catalyzing a rate-limiting reaction. Heme also affects translation of the enzyme and its transfer from the cytosol to the mitochondrion.
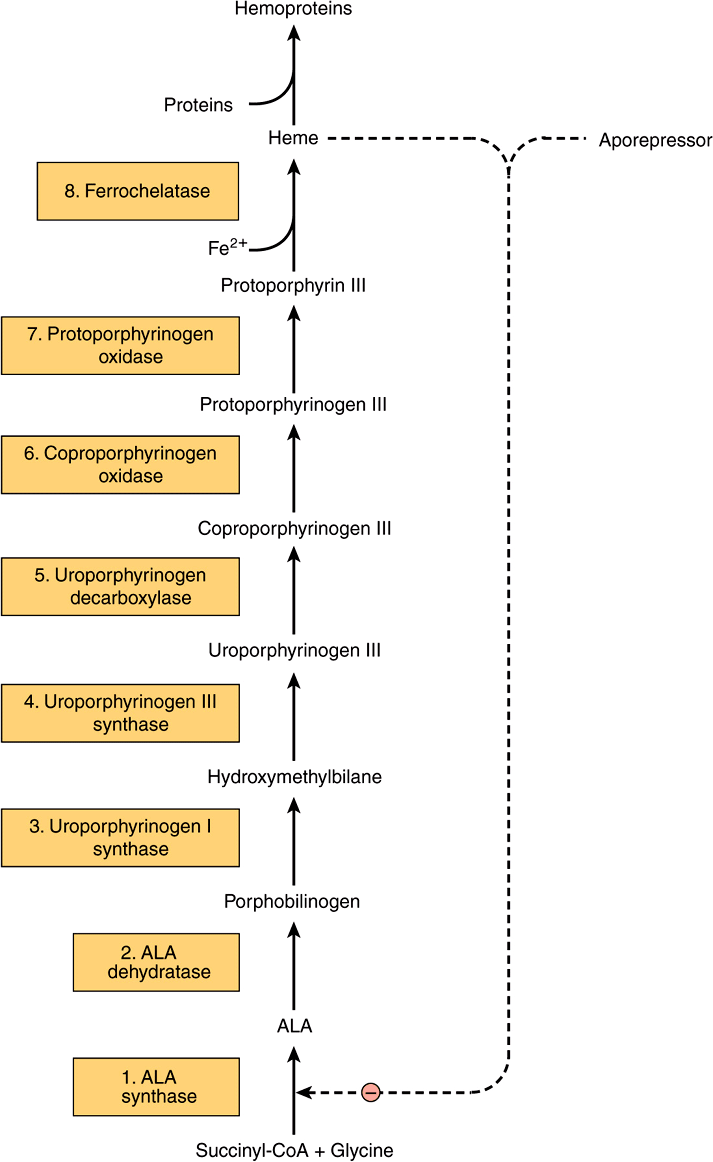
FIGURE 31–9 Intermediates, enzymes, and regulation of heme synthesis. The enzyme numbers are those referred to in column 1 of Table 31-2. Enzymes 1, 6, 7, and 8 are located in mitochondria, the others in the cytosol. Mutations in the gene encoding enzyme 1 cause X-linked sideroblastic anemia. Mutations in the genes encoding enzymes 2-8 cause the porphyrias, though only a few cases due to deficiency of enzyme 2 have been reported. Regulation of hepatic heme synthesis occurs at ALA synthase (ALAS1) by a repressionderepression mechanism mediated by heme and its hypothetical aporepressor. The dotted lines indicate the negative (–) regulation by repression. Enzyme 3 is also called porphobilinogen deaminase or hydroxymethylbilane synthase.
TABLE 31–2 Summary of Major Findings in the Porphyrias 1
Many drugs when administered to humans can result in a marked increase in ALAS1. Most of these drugs are metabolized by a system in the liver that utilizes a specific hemo-protein, cytochrome P450 (see Chapter 53). During their metabolism, the utilization of heme by cytochrome P450 is greatly increased, which in turn diminishes the intracellular heme concentration. This latter event effects a derepression of ALAS1 with a corresponding increased rate of heme synthesis to meet the needs of the cells.
Several factors affect drug-mediated derepression of ALAS1 in the liver—eg, the administration of glucose can prevent it, as can the administration of hematin (an oxidized form of heme).
The importance of some of these regulatory mechanisms is further discussed below when the porphyrias are described.
Regulation of the erythroid form of ALAS (ALAS2) differs from that of ALAS1. For instance, it is not induced by the drugs that affect ALAS1, and it does not undergo feedback regulation by heme.
PORPHYRINS ARE COLORED & FLUORESCE
The various porphyrinogens are colorless, whereas the various porphyrins are all colored. In the study of porphyrins or porphyrin derivatives, the characteristic absorption spectrum that each exhibits—in both the visible and the ultraviolet regions of the spectrum—is of great value. An example is the absorption curve for a solution of porphyrin in 5% hydrochloric acid (Figure 31–10). Note particularly the sharp absorption band near 400 nm. This is a distinguishing feature of the porphyrin ring and is characteristic of all porphyrins regardless of the side chains present. This band is termed the Soret band after its discoverer, the French physicist Charles Soret.
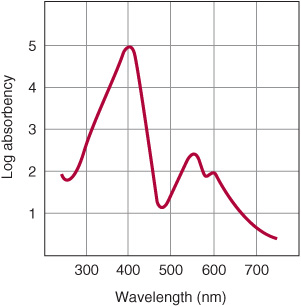
FIGURE 31–10 Absorption spectrum of hematoporphyrin (0.01% solution in 5% HCl).
When porphyrins dissolved in strong mineral acids or in organic solvents are illuminated by ultraviolet light, they emit a strong red fluorescence. This fluorescence is so characteristic that it is often used to detect small amounts of free porphyrins. The double bonds joining the pyrrole rings in the porphyrins are responsible for the characteristic absorption and fluorescence of these compounds; these double bonds are absent in the porphyrinogens.
An interesting application of the photodynamic properties of porphyrins is their possible use in the treatment of certain types of cancer, a procedure called cancer phototherapy. Tumors often take up more porphyrins than do normal tissues. Thus, hematoporphyrin or other related compounds are administered to a patient with an appropriate tumor. The tumor is then exposed to an argon laser, which excites the porphyrins, producing cytotoxic effects.
Spectrophotometry Is Used to Test for Porphyrins & Their Precursors
Coproporphyrins and uroporphyrins are of clinical interest because they are excreted in increased amounts in the porphyrias. These compounds, when present in urine or feces, can be separated from each other by extraction with appropriate solvent mixtures. They can then be identified and quantified using spectrophotometric methods.
ALA and PBG can also be measured in urine by appropriate colorimetric tests.
THE PORPHYRIAS ARE GENETIC DISORDERS OF HEME METABOLISM
The porphyrias are a group of disorders due to abnormalities in the pathway of biosynthesis of heme; they can be genetic or acquired. They are not prevalent, but it is important to consider them in certain circumstances (eg, in the differential diagnosis of abdominal pain and of a variety of neuropsychiatric findings); otherwise, patients will be subjected to inappropriate treatments. It has been speculated that King George III had a type of porphyria, which may account for his periodic confinements in Windsor Castle and perhaps for some of his views regarding American colonists. Also, the photosensitivity (favoring nocturnal activities) and severe disfigurement exhibited by some victims of congenital erythropoietic porphyria have led to the suggestion that these individuals may have been the prototypes of so-called were-wolves. No evidence to support this notion has been adduced.
Biochemistry Underlies the Causes, Diagnoses, & Treatments of the Porphyrias
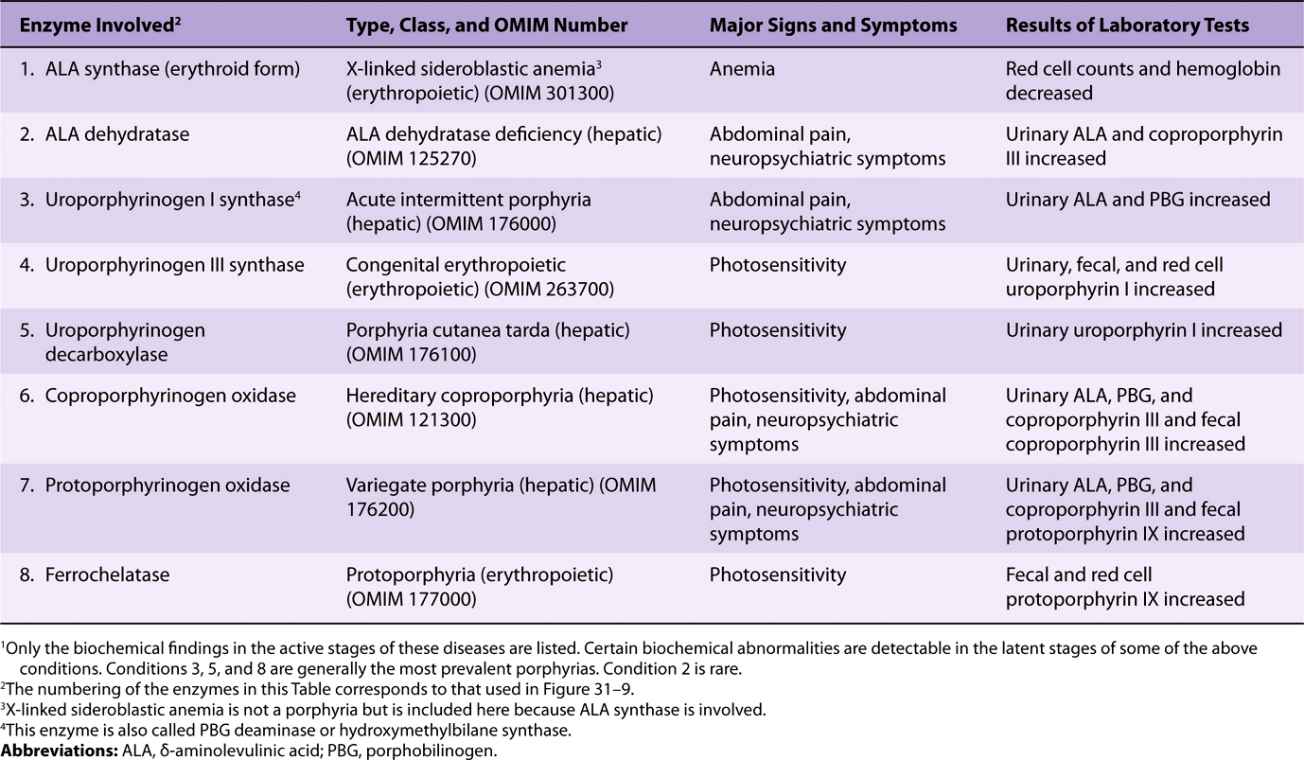
Six major types of porphyria have been described, resulting from depressions in the activities of enzymes 3 through 8 shown in Figure 31–9 (see also Table 31-2). Assay of the activity of one or more of these enzymes using an appropriate source (eg, red blood cells) is thus important in making a definitive diagnosis in a suspected case of porphyria. Individuals with low activities of enzyme 1 (ALAS2) develop anemia, not porphyria (see Table 31-2). Patients with low activities of enzyme 2 (ALA dehydratase) have been reported, but very rarely; the resulting condition is called ALA dehydratase-deficient porphyria.
In general, the porphyrias described are inherited in an autosomal dominant manner, with the exception of congenital erythropoietic porphyria, which is inherited in a recessive mode. The precise abnormalities in the genes directing synthesis of the enzymes involved in heme biosynthesis have been determined in some instances. Thus, the use of appropriate gene probes has made possible the prenatal diagnosis of some of the porphyrias.
As is true of most inborn errors, the signs and symptoms of porphyria result from either a deficiency of metabolic products beyond the enzymatic block or from an accumulation of metabolites behind the block.
If the enzyme lesion occurs early in the pathway prior to the formation of porphyrinogens (eg, enzyme 3 of Figure 31–9, which is affected in acute intermittent porphyria), ALA and PBG will accumulate in body tissues and fluids (Figure 31–11). Clinically, patients complain of abdominal pain and neuropsychiatric symptoms. The precise biochemical cause of these symptoms has not been determined but may relate to elevated levels of ALA or PBG or to a deficiency of heme.
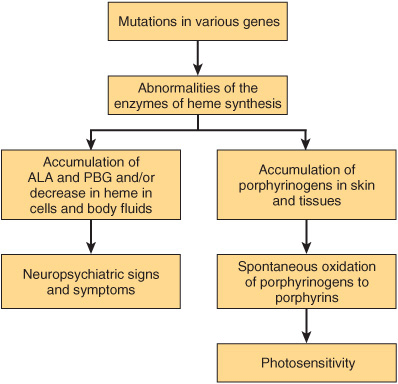
FIGURE 31–11 Biochemical causes of the major signs and symptoms of the porphyrias.
On the other hand, enzyme blocks later in the pathway result in the accumulation of the porphyrinogens indicated in Figures 31-9 and 31-11. Their oxidation products, the corresponding porphyrin derivatives, cause photosensitivity, a reaction to visible light of about 400 nm. The porphyrins, when exposed to light of this wavelength, are thought to become “excited” and then react with molecular oxygen to form oxygen radicals. These latter species injure lysosomes and other organelles. Damaged lysosomes release their degradative enzymes, causing variable degrees of skin damage, including scarring.
The porphyrias can be classified on the basis of the organs or cells that are most affected. These are generally organs or cells in which synthesis of heme is particularly active. The bone marrow synthesizes considerable hemoglobin, and the liver is active in the synthesis of another hemoprotein, cytochrome P450. Thus, one classification of the porphyrias is to designate them as predominantly either erythropoietic or hepatic; the types of porphyrias that fall into these two classes are so characterized in Table 31-2. Porphyrias can also be classified as acute or cutaneous on the basis of their clinical features. Why do specific types of porphyria affect certain organs more markedly than others? A partial answer is that the levels of metabolites that cause damage (eg, ALA, PBG, specific porphyrins, or lack of heme) can vary markedly in different organs or cells depending upon the differing activities of their heme-forming enzymes.
As described above, ALAS1 is the key regulatory enzyme of the heme biosynthetic pathway in liver. A large number of drugs (eg, barbiturates, griseofulvin) induce the enzyme. Most of these drugs do so by inducing cytochrome P450 (see Chapter 53), which uses up heme and thus derepresses (induces) ALAS1. In patients with porphyria, increased activities of ALAS1 result in increased levels of potentially harmful heme precursors prior to the metabolic block. Thus, taking drugs that cause induction of cytochrome P450 (so-called microsomal inducers) can precipitate attacks of porphyria.
The diagnosis of a specific type of porphyria can generally be established by consideration of the clinical and family history, the physical examination, and appropriate laboratory tests. The major findings in the six principal types of porphyria are listed in Table 31-2.
High levels of lead can affect heme metabolism by combining with SH groups in enzymes such as ferrochelatase and ALA dehydratase. This affects porphyrin metabolism. Elevated levels of protoporphyrin are found in red blood cells, and elevated levels of ALA and of coproporphyrin are found in urine.
It is hoped that treatment of the porphyrias at the gene level will become possible. In the meantime, treatment is essentially symptomatic. It is important for patients to avoid drugs that cause induction of cytochrome P450. Ingestion of large amounts of carbohydrates (glucose loading) or administration of hematin (a hydroxide of heme) may repress ALAS1, resulting in diminished production of harmful heme precursors. Patients exhibiting photosensitivity may benefit from administration of β-carotene; this compound appears to lessen production of free radicals, thus diminishing photosensitivity. Sunscreens that filter out visible light can also be helpful to such patients.
CATABOLISM OF HEME PRODUCES BILIRUBIN
Under normal conditions in human adults, some 200 billion erythrocytes are destroyed per day. Thus, a 70-kg human turns over approximately 6 g of hemoglobin daily. When hemoglobin is destroyed in the body, globin is degraded to its constituent amino acids, which are reused, and the iron of heme enters the iron pool, also for reuse. The iron-free porphyrin portion of heme is also degraded, mainly in the reticuloendothelial cells of the liver, spleen, and bone marrow.
The catabolism of heme from all of the heme proteins appears to be carried out in the microsomal fractions of cells by a complex enzyme system called heme oxygenase. By the time the heme derived from heme proteins reaches the oxygenase system, the iron has usually been oxidized to the ferric form, constituting hemin. The heme oxygenase system is substrate-inducible. As depicted in Figure 31–12, the hemin is reduced to heme with NADPH, and, with the help of more NADPH, oxygen is added to the α-methyne bridge between pyrroles I and II of the porphyrin. The ferrous iron is again oxidized to the ferric form. With the further addition of oxygen, ferric ion is released, carbon monoxide is produced, and an equimolar quantity of biliverdin results from the splitting of the tetrapyrrole ring.
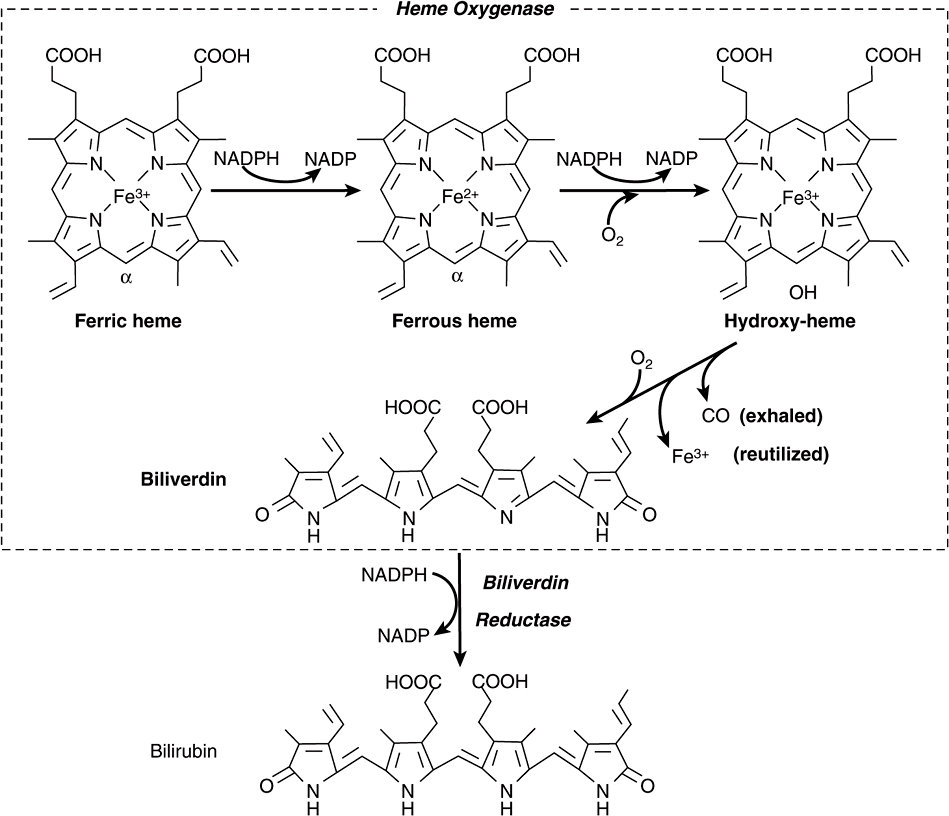
FIGURE 31–12 Schematic representation of the microsomal heme oxygenase system. The order of side-chain groups in bilirubin (from left to right) is M, V, M, P, P, M, M, V (M, methyl; V, vinyl; P, propionyl). (Redrawn, with permission, by Antony McDonagh from Schmid R, McDonough AF in: The Porphyrins. Dolphin D [ed.]. Academic Press, 1978. Reprinted with permission from Elsevier.)
In birds and amphibia, the green biliverdin IX is excreted; in mammals, a soluble enzyme called biliverdin reductase reduces the methyne bridge between pyrrole III and pyrrole IV to a methylene group to produce bilirubin, a yellow pigment (Figure 31–12).
It is estimated that 1 g of hemoglobin yields 35 mg of bilirubin. The daily bilirubin formation in human adults is approximately 250-350 mg, deriving mainly not only from hemoglobin, but from ineffective erythropoiesis and from various other heme proteins such as cytochrome P450.
The chemical conversion of heme to bilirubin by reticuloendothelial cells can be observed in vivo as the purple color of the heme in a hematoma is slowly converted to the yellow pigment of bilirubin.
Bilirubin formed in peripheral tissues is transported to the liver by plasma albumin. The further metabolism of bilirubin occurs primarily in the liver. It can be divided into three processes. (1) uptake of bilirubin by liver parenchymal cells, (2) conjugation of bilirubin with glucuronate in the endoplasmic reticulum, and (3) secretion of conjugated bilirubin into the bile. Each of these processes will be considered separately.
THE LIVER TAKES UP BILIRUBIN
Bilirubin is only sparingly soluble in water, but its solubility in plasma is increased by noncovalent binding to albumin. Each molecule of albumin appears to have one high-affinity site and one low-affinity site for bilirubin. In 100 mL of plasma, approximately 25 mg of bilirubin can be tightly bound to albumin at its high-affinity site. Bilirubin in excess of this quantity can be bound only loosely and thus can easily be detached and diffuse into tissues. A number of compounds such as antibiotics and other drugs compete with bilirubin for the high-affinity binding site on albumin. Thus, these compounds can displace bilirubin from albumin and have significant clinical effects.
In the liver, the bilirubin is removed from albumin and taken up at the sinusoidal surface of the hepatocytes by a carrier-mediated saturable system. This facilitated transport system has a very large capacity, so that even under pathologic conditions the system does not appear to be rate limiting in the metabolism of bilirubin.
Since this facilitated transport system allows the equilibrium of bilirubin across the sinusoidal membrane of the hepatocyte, the net uptake of bilirubin will be dependent upon the removal of bilirubin via subsequent metabolic pathways.
Once bilirubin enters the hepatocytes, it can bind to certain cytosolic proteins, which help to keep it solubilized prior to conjugation. Ligandin (a member of the family of glutathione S-transferases) and protein Y are the involved proteins. They may also help to prevent efflux of bilirubin back into the blood stream.
Conjugation of Bilirubin with Glucuronic Acid Occurs in the Liver
Bilirubin is nonpolar and would persist in cells (eg, bound to lipids) if not rendered water-soluble. Hepatocytes convert bilirubin to a polar form, which is readily excreted in the bile, by adding glucuronic acid molecules to it. This process is called conjugation and can employ polar molecules other than glucuronic acid (eg, sulfate). Many steroid hormones and drugs are also converted to water-soluble derivatives by conjugation in preparation for excretion (see Chapter 53).
The conjugation of bilirubin is catalyzed by a specific glucuronosyltransferase. The enzyme is mainly located in the endoplasmic reticulum, uses UDP-glucuronic acid as the glucuronosyl donor, and is referred to as bilirubin-UGT. Bilirubin monoglucuronide is an intermediate and is subsequently converted to the diglucuronide (Figures 31-13 and 31-14). Most of the bilirubin excreted in the bile of mammals is in the form of bilirubin diglucuronide. However, when bilirubin conjugates exist abnormally in human plasma (eg, in obstrucive jaundice), they are predominantly monoglucuronides. Bilirubin-UGT activity can be induced by a number of clinically useful drugs, including phenobarbital. More information about glucuronosylation is presented below in the discussion of inherited disorders of bilirubin conjugation.
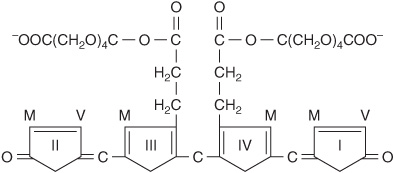
FIGURE 31–13 Structure of bilirubin diglucuronide (conjugated, “direct-reacting” bilirubin). Glucuronic acid is attached via ester linkage to the two propionic acid groups of bilirubin to form an acylglucuronide.
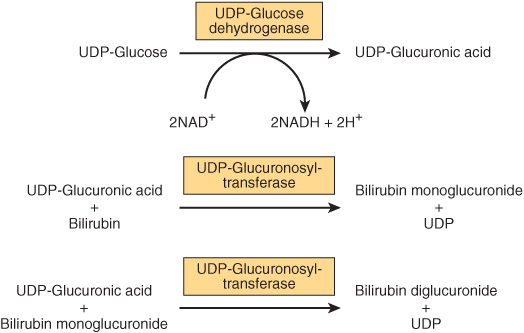
FIGURE 31–14 Conjugation of bilirubin with glucuronic acid. The glucuronate donor, UDP-glucuronic acid, is formed from UDP-glucose as depicted. The UDP-glucuronosyltransferase is also called bilirubin-UGT.
Bilirubin Is Secreted into Bile
Secretion of conjugated bilirubin into the bile occurs by an active transport mechanism, which is probably rate-limiting for the entire process of hepatic bilirubin metabolism. The protein involved is MRP-2 (multidrug-resistance-like protein 2), also called multispecific organic anion transporter (MOAT). It is located in the plasma membrane of the bile canalicular membrane and handles a number of organic anions. It is a member of the family of ATP-binding cassette transporters. The hepatic transport of conjugated bilirubin into the bile is inducible by those same drugs that are capable of inducing the conjugation of bilirubin. Thus, the conjugation and excretion systems for bilirubin behave as a coordinated functional unit.
Figure 31–15 summarizes the three major processes involved in the transfer of bilirubin from blood to bile. Sites that are affected in a number of conditions causing jaundice (see below) are also indicated.
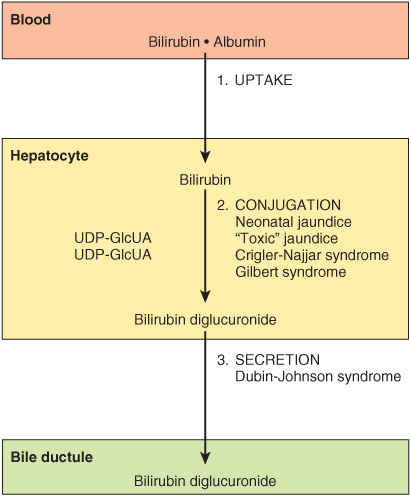
FIGURE 31–15 Diagrammatic representation of the three major processes (uptake, conjugation, and secretion) involved in the transfer of bilirubin from blood to bile. Certain proteins of hepatocytes, such as ligandin (a member of the glutathione S-transferase family of enzymes) and Y protein, bind intracellular bilirubin and may prevent its efflux into the blood stream. The process affected in a number of conditions causing jaundice is also shown.
Conjugated Bilirubin Is Reduced to Urobilinogen by Intestinal Bacteria
As the conjugated bilirubin reaches the terminal ileum and the large intestine, the glucuronides are removed by specific bacterial enzymes (β-glucuronidases), and the pigment is subsequently reduced by the fecal flora to a group of colorless tetrapyrrolic compounds called urobilinogens. In the terminal ileum and large intestine, a small fraction of the urobilinogens is reabsorbed and reexcreted through the liver to constitute the enterohepatic urobilinogen cycle. Under abnormal conditions, particularly when excessive bile pigment is formed or liver disease interferes with this intrahepatic cycle, urobilinogen may also be excreted in the urine.
Normally, most of the colorless urobilinogens formed in the colon by the fecal flora are oxidized there to urobilins (colored compounds) and are excreted in the feces. Darkening of feces upon standing in air is due to the oxidation of residual urobilinogens to urobilins.
HYPERBILIRUBINEMIA CAUSES JAUNDICE
When bilirubin in the blood exceeds 1 mg/dL (17.1 μmol/L), hyperbilirubinemia exists. Hyperbilirubinemia may be due to the production of more bilirubin than the normal liver can excrete, or it may result from the failure of a damaged liver to excrete bilirubin produced in normal amounts. In the absence of hepatic damage, obstruction of the excretory ducts of the liver—by preventing the excretion of bilirubin—will also cause hyperbilirubinemia. In all these situations, bilirubin accumulates in the blood, and when it reaches a certain concentration (approximately 2-2.5 mg/dL), it diffuses into the tissues, which then become yellow. That condition is called jaundice or icterus.
In clinical studies of jaundice, measurement of bilirubin in the serum is of a great value. A method for quantitatively assaying the bilirubin content of the serum was first devised by van den Bergh by application of Ehrlich’s test for bilirubin in urine. The Ehrlich reaction is based on the coupling of diazotized sulfanilic acid (Ehrlich’s diazo reagent) and bilirubin to produce a reddish-purple azo compound. In the original procedure as described by Ehrlich, methanol was used to provide a solution in which both bilirubin and the diazo regent were soluble. van den Bergh inadvertently omitted the methanol on an occasion when assay of bile pigment in human bile was being attempted. To his surprise, normal development of the color occurred “directly.” This form of bilirubin that would react without the addition of methanol was thus termed “direct-reacting.” It was then found that this same direct reaction would also occur in serum from cases of jaundice due to biliary obstruction. However, it was still necessary to add methanol to detect bilirubin in normal serum or the one that was present in excess in serum from cases of hemolytic jaundice where no evidence of obstruction was to be found. To that form of bilirubin, which could be measured only after the addition of methanol, the term “indirect-reacting” was applied.
It was subsequently discovered that the indirect bilirubin is “free” (unconjugated) bilirubin en route to the liver from the reticuloendothelial tissues, where the bilirubin was originally produced by the breakdown of heme porphyrins. Since this bilirubin is not water-soluble, it requires methanol to initiate coupling with the diazo reagent. In the liver, the free bilirubin becomes conjugated with glucuronic acid, and the conjugate, predominantly bilirubin diglucuronide, can then be excreted into the bile. Furthermore, conjugated bilirubin, being water-soluble, can react directly with the diazo reagent, so that the “direct bilirubin” of van den Bergh is actually a bilirubin conjugate (bilirubin glucuronide).
Depending on the type of bilirubin present in plasma—ie, unconjugated or conjugated—hyperbilirubinemia may be classified as retention hyperbilirubinemia, due to overproduction, or regurgitation hyperbilirubinemia, due to reflux into the bloodstream because of biliary obstruction.
Separation and quantitation of unconjugated bilirubin and the conjugated species can be performed using high-pressure liquid chromatography.
Because of its hydrophobicity, only unconjugated bilirubin can cross the blood-brain barrier into the central nervous system; thus, encephalopathy due to hyperbilirubinemia (kernicterus) can occur only in connection with unconjugated bilirubin, as found in retention hyperbilirubinemia. On the other hand, because of its water-solubility, only conjugated bilirubin can appear in urine. Accordingly, choluric jaundice (choluria is the presence of bile pigments in the urine) occurs only in regurgitation hyperbilirubinemia, and acholuric jaundice occurs only in the presence of an excess of unconjugated bilirubin.
Table 31-3 lists some causes of unconjugated and conjugated hyperbilirubinemia. These conditions are briefly described in the following sections.
TABLE 31–3 Some Causes of Unconjugated and Conjugated Hyperbilirubinemia
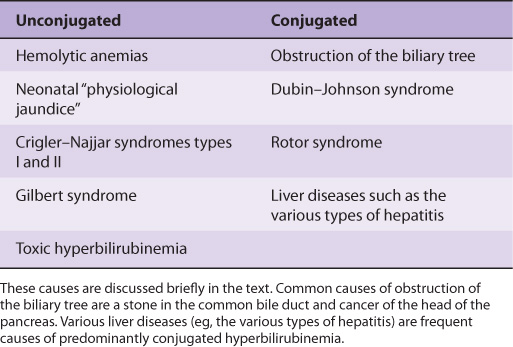
Elevated Amounts of Unconjugated Bilirubin in Blood Occur in a Number of Conditions
Hemolytic Anemias
Hemolytic anemias are important causes of unconjugated hyperbilirubinemia, though unconjugated hyperbilirubinemia is usually only slight (<4 mg/dL; <68.4 μmol/L) even in the event of extensive hemolysis because of the healthy liver’s large capacity for handling bilirubin.
Neonatal “Physiologic Jaundice”
This transient condition is the most common cause of unconjugated hyperbilirubinemia. It results from an accelerated hemolysis around the time of birth and an immature hepatic system for the uptake, conjugation, and secretion of bilirubin. Not only is the bilirubin-UGT activity reduced, but there probably is reduced synthesis of the substrate for that enzyme, UDP-glucuronic acid. Since the increased amount of bilirubin is unconjugated, it is capable of penetrating the blood-brain barrier when its concentration in plasma exceeds that which can be tightly bound by albumin (20-25 mg/dL). This can result in a hyperbilirubinemic toxic encephalopathy, or kernicterus, which can cause mental retardation. Because of the recognized inducibility of this bilirubin-metabolizing system, phenobarbital has been administered to jaundiced neonates and is effective in this disorder. In addition, exposure to blue light (phototherapy) promotes the hepatic excretion of unconjugated bilirubin by converting some of the bilirubin to other derivatives such as maleimide fragments and geometric isomers that are excreted in the bile.
Crigler-Najjar Syndrome, Type I; Congenital Nonhemolytic Jaundice
The type-I Crigler-Najjar syndrome is a rare autosomal recessive disorder. It is characterized by severe congenital jaundice (serum bilirubin usually exceeds 20 mg/dL) due to mutations in the gene encoding bilirubin-UGT activity in hepatic tissues. The disease is often fatal within the first 15 months of life. Children with this condition have been treated with phototherapy, resulting in some reduction in plasma bilirubin levels. Phenobarbital has no effect on the formation of bilirubin glucuronides in patients with the type I Crigler-Najjar syndrome. A liver transplant may be curative.
It should be noted that the gene encoding human bilirubin-UGT is part of a large UGT gene complex situated on chromosome 2. Many different substrates are subjected to glucuronosylation; therefore, many glucuronosyltransferases are required. The complex contains some 13 substrate-specific first exons, each with its own promoter. Four are pseudogenes, so nine different isoforms with differing glucuronosyltransferase activities are encoded. Exon A1 is that involved with conjugation of bilirubin. In the case of bilirubin, exon A1 is spliced to DNA containing exons 2-5, producing bilirubin-UGT. Other transferases are produced by splicing other first exons (members of A 2-13) to exons 2-5.
Crigler-Najjar Syndrome, Type II
This rare inherited disorder also results from mutations in the gene encoding bilirubin-UGT, but some activity of the enzyme is retained and the condition has a more benign course than type I. Serum bilirubin concentrations usually do not exceed 20 mg/dL. Patients with this condition can respond to treatment with large doses of phenobarbital.
Gilbert Syndrome
Again, this relatively prevalent condition is caused by mutations in the gene encoding bilirubin-UGT. It is more common among males. Approximately 30% of the enzyme’s activity is preserved and the condition is entirely harmless.
Toxic Hyperbilirubinemia
Unconjugated hyperbilirubinemia can result from toxin-induced liver dysfunction such as that caused by chloroform, arsphenamines, carbon tetrachloride, acetaminophen, hepatitis virus, cirrhosis, and Amanita mushroom poisoning. These acquired disorders are due to hepatic parenchymal cell damage, which impairs conjugation.
Obstruction in the Biliary Tree Is the Most Common Cause of Conjugated Hyperbilirubinemia
Obstruction of the Biliary Tree
Conjugated hyperbilirubinemia commonly results from blockage of the hepatic or common bile ducts, most often due to a gallstone or to cancer of the head of the pancreas (Figure 31–16). Because of the obstruction, bilirubin diglucuronide cannot be excreted. It thus regurgitates into the hepatic veins and lymphatics, and conjugated bilirubin appears in the blood and urine (choluric jaundice). Also, the stools are usually pale in color, and should be examined routinely in any case of jaundice.
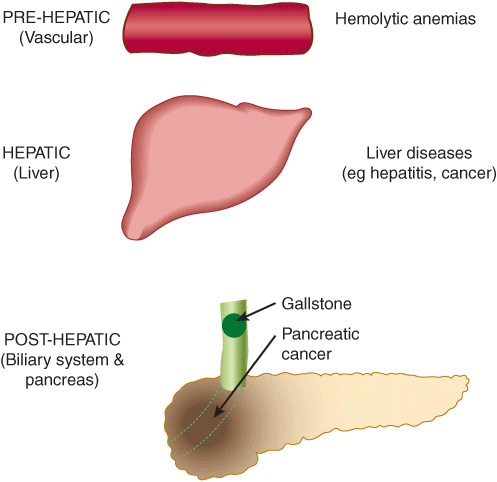
FIGURE 31–16 Diagrammatic representation of some major causes of jaundice. Prehepatic indicates events in the blood stream; the major cause would be various forms of hemolytic anemia (see Chapter 52). Hepatic signifies events in the liver, such as the various types of hepatitis or other forms of liver disease (eg, cancer). Posthepatic refers to events in the biliary tree; the major causes of posthepatic jaundice are obstruction of the common bile duct by a gallstone (biliary calculus) or by cancer of the head of the pancreas.
The term cholestatic jaundice is used to include all cases of extrahepatic obstructive jaundice. It also covers those cases of jaundice that exhibit conjugated hyperbilirubinemia due to micro-obstruction of intrahepatic biliary ductules by swollen, damaged hepatocytes (eg, as may occur in infectious hepatitis).
This benign autosomal recessive disorder consists of conjugated hyperbilirubinemia in childhood or during adult life. The hyperbilirubinemia is caused by mutations in the gene encoding MRP-2 (see above), the protein involved in the secretion of conjugated bilirubin into bile. The centrilobular hepatocytes contain an abnormal black pigment that may be derived from epinephrine.
Rotor Syndrome
This is a rare benign condition characterized by chronic conjugated hyperbilirubinemia and normal liver histology. Its precise cause has not been identified.
Some Conjugated Bilirubin Can Bind Covalently to Albumin
When levels of conjugated bilirubin remain high in plasma, a fraction can bind covalently to albumin (δ [delta] bilirubin). Because it is bound covalently to albumin, this fraction has a longer half-life in plasma than does conventional conjugated bilirubin. Thus, it remains elevated during the recovery phase of obstructive jaundice after the remainder of the conjugated bilirubin has declined to normal levels; this explains why some patients continue to appear jaundiced after conjugated bilirubin levels have returned to normal.
Urobilinogen & Bilirubin in Urine Are Clinical Indicators
Normally, there are mere traces of urobilinogen in the urine. In complete obstruction of the bile duct, no urobilinogen is found in the urine, since bilirubin has no access to the intestine, where it can be converted to urobilinogen. In this case, the presence of bilirubin (conjugated) in the urine without urobilinogen suggests obstructive jaundice, either intrahepatic or posthepatic.
In jaundice secondary to hemolysis, the increased production of bilirubin leads to increased production of urobilinogen, which appears in the urine in large amounts. Bilirubin is not usually found in the urine in hemolytic jaundice (because unconjugated bilirubin does not pass into the urine), so that the combination of increased urobilinogen and absence of bilirubin is suggestive of hemolytic jaundice. Increased blood destruction from any cause brings about an increase in urine urobilinogen.
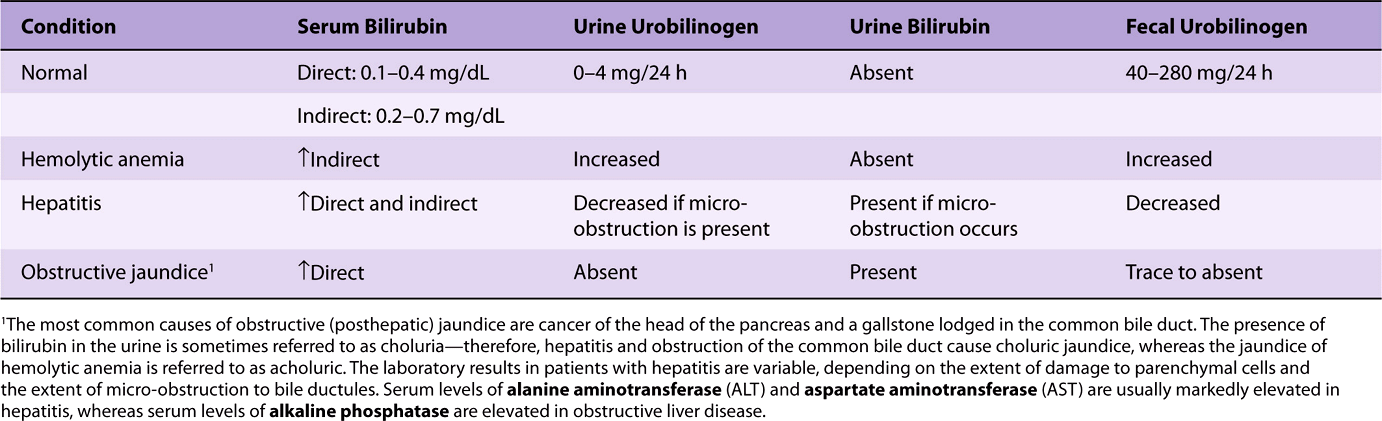
Table 31-4 summarizes laboratory results obtained on patients with three different causes of jaundice—hemolytic anemia (a prehepatic cause), hepatitis (a hepatic cause), and obstruction of the common bile duct (a posthepatic cause) (see Figure 31–16). Laboratory tests on blood (evaluation of the possibility of a hemolytic anemia and measurement of prothrombin time) and on serum (eg, electrophoresis of proteins; activities of the enzymes ALT, AST, and alkaline phosphatase) are also important in helping to distinguish between prehepatic, hepatic, and posthepatic causes of jaundice.
TABLE 31–4 Laboratory Results in Normal Patients and Patients with Three Different Causes of Jaundice
SUMMARY
![]() Hemoproteins, such as hemoglobin and the cytochromes, contain heme. Heme is an iron-porphyrin compound (Fe2+-protoporphyrin IX) in which four pyrrole rings are joined by methyne bridges. The eight side groups (methyl, vinyl, and propionyl substituents) on the four pyrrole rings of heme are arranged in a specific sequence.
Hemoproteins, such as hemoglobin and the cytochromes, contain heme. Heme is an iron-porphyrin compound (Fe2+-protoporphyrin IX) in which four pyrrole rings are joined by methyne bridges. The eight side groups (methyl, vinyl, and propionyl substituents) on the four pyrrole rings of heme are arranged in a specific sequence.
![]() Biosynthesis of the heme ring occurs in mitochondria and cytosol via eight enzymatic steps. It commences with formation of δ-aminolevulinate from succinyl-CoA and glycine in a reaction catalyzed by the ALA synthase, the regulatory enzyme of the pathway.
Biosynthesis of the heme ring occurs in mitochondria and cytosol via eight enzymatic steps. It commences with formation of δ-aminolevulinate from succinyl-CoA and glycine in a reaction catalyzed by the ALA synthase, the regulatory enzyme of the pathway.
![]() Genetically determined abnormalities of seven of the eight enzymes involved in heme biosynthesis result in the inherited porphyrias. Red blood cells and liver are the major sites of metabolic expression of the porphyrias. Photosensitivity and neurologic problems are common complaints. Intake of certain compounds (such as lead) can cause acquired porphyrias. Increased amounts of porphyrins or their precursors can be detected in blood and urine, facilitating diagnosis.
Genetically determined abnormalities of seven of the eight enzymes involved in heme biosynthesis result in the inherited porphyrias. Red blood cells and liver are the major sites of metabolic expression of the porphyrias. Photosensitivity and neurologic problems are common complaints. Intake of certain compounds (such as lead) can cause acquired porphyrias. Increased amounts of porphyrins or their precursors can be detected in blood and urine, facilitating diagnosis.
![]() Catabolism of the heme ring is initiated by the enzyme heme oxygenase, producing a linear tetrapyrrole.
Catabolism of the heme ring is initiated by the enzyme heme oxygenase, producing a linear tetrapyrrole.
![]() Biliverdin is an early product of catabolism and on reduction yields bilirubin. The latter is transported by albumin from peripheral tissues to the liver, where it is taken up by hepatocytes. The iron of heme and the amino acids of globin are conserved and reutilized.
Biliverdin is an early product of catabolism and on reduction yields bilirubin. The latter is transported by albumin from peripheral tissues to the liver, where it is taken up by hepatocytes. The iron of heme and the amino acids of globin are conserved and reutilized.
![]() In the liver, bilirubin is made water-soluble by conjugation with two molecules of glucuronic acid and is secreted into the bile. The action of bacterial enzymes in the gut produces urobilinogen and urobilin, which are excreted in the feces and urine.
In the liver, bilirubin is made water-soluble by conjugation with two molecules of glucuronic acid and is secreted into the bile. The action of bacterial enzymes in the gut produces urobilinogen and urobilin, which are excreted in the feces and urine.
![]() Jaundice is due to elevation of the level of bilirubin in the blood. The causes of jaundice can be classified as prehepatic (eg, hemolytic anemias), hepatic (eg, hepatitis), and posthepatic (eg, obstruction of the common bile duct). Measurements of plasma total and nonconjugated bilirubin, of urinary urobilinogen and bilirubin, and of certain serum enzymes as well as inspection and analysis of stool samples help distinguish between these causes.
Jaundice is due to elevation of the level of bilirubin in the blood. The causes of jaundice can be classified as prehepatic (eg, hemolytic anemias), hepatic (eg, hepatitis), and posthepatic (eg, obstruction of the common bile duct). Measurements of plasma total and nonconjugated bilirubin, of urinary urobilinogen and bilirubin, and of certain serum enzymes as well as inspection and analysis of stool samples help distinguish between these causes.
REFERENCES
Beckett G, Walker S, Rae T, Ashby P: Liver disease (Chapter 13). In: Lecture Notes: Clinical Biochemistry. 8th ed. Wiley-Blackwell, 2010.
Deacon AC, Whatley SD, Elder GH: Porphyrins and disorders of porphyrin metabolism. In: Tietz Textbook of Clinical Chemistry and Molecular Diagnostics, 4th ed. Ch. 32. Burtis CA, Ashwood ER, Bruns DE (editors). Elsevier Saunders, 2006.
Desnick RJ, Astrin KH: The porphyrias. In: Harrison’s Principles of Internal Medicine, 17th ed. Fauci AS et al (editors). Ch. 352. McGraw-Hill, 2008.
Dufour DR: Liver disease. In: Tietz Textbook of Clinical Chemistry and Molecular Diagnostics, 4th ed. Burtis CA, Ashwood ER, Bruns DE (editors). Ch. 47. Elsevier Saunders, 2006.
Higgins T, Beutler E, Doumas BT: Hemoglobin, iron and bilirubin. In: Tietz Textbook of Clinical Chemistry and Molecular Diagnostics, 4th ed. Burtis CA, Ashwood ER, Bruns DE (editors). Ch. 31. Elsevier Saunders, 2006.
Pratt DS, Kaplan MM: Evaluation of liver function. In: Harrison’s Principles of Internal Medicine, 17th ed. Fauci AS et al (editors). Ch. 296. McGraw-Hill, 2008.
Pratt DS, Kaplan MM: Jaundice. In: Harrison’s Principles of Internal Medicine, 17th ed. Fauci AS et al (editors). Ch. 43. McGraw-Hill, 2008.
Wolkoff AW: The hyperbilirubinemias. In: Harrison’s Principles of Internal Medicine, 17th ed. Fauci AS et al (editors). Ch. 297. McGraw-Hill, 2008.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


