Etiology In at least 90% of patients with Cushing’s disease, ACTH excess is caused by a corticotrope pituitary microadenoma, often only a few millimeters in diameter. Pituitary macroadenomas (i.e., tumors >1 cm in size) are found in only 5–10% of patients. Pituitary corticotrope adenomas usually occur sporadically but very rarely can be found in the context of multiple endocrine neoplasia type 1 (MEN 1) (Chap. 408).
Ectopic ACTH production is predominantly caused by occult carcinoid tumors, most frequently in the lung, but also in thymus or pancreas. Because of their small size, these tumors are often difficult to locate. Advanced small-cell lung cancer can cause ectopic ACTH production. In rare cases, ectopic CRH and/or ACTH production has been found to originate from medullary thyroid carcinoma or pheochromocytoma, the latter co-secreting catecholamines and ACTH.
The majority of patients with ACTH-independent cortisol excess harbor a cortisol-producing adrenal adenoma; intratumor mutations, i.e., somatic mutations in the PKA catalytic subunit PRKACA, have been identified as cause of disease in 40% of these tumors. Adrenocortical carcinomas may also cause ACTH-independent disease and are often large, with excess production of several corticosteroid classes.
A rare but notable cause of adrenal cortisol excess is macronodular adrenal hyperplasia with low circulating ACTH, but with evidence for autocrine stimulation of cortisol production via intraadrenal ACTH production. These hyperplastic nodules are often also characterized by ectopic expression of G protein–coupled receptors not usually found in the adrenal, including receptors for luteinizing hormone, vasopressin, serotonin, interleukin 1, catecholamines, or gastric inhibitory peptide (GIP), the cause of food-dependent Cushing’s. Activation of these receptors results in upregulation of PKA signaling, as physiologically occurs with ACTH, with a subsequent increase in cortisol production. A combination of germline and somatic mutations in the tumor-suppressor gene ARMC5 have been identified as a prevalent cause of Cushing’s due to macronodular adrenal hyperplasia. Germline mutations in the PKA catalytic subunit PRKACA can represent a rare cause of macronodular adrenal hyperplasia associated with cortisol excess.
Mutations in one of the regulatory subunits of PKA, PRKAR1A, are found in patients with primary pigmented nodular adrenal disease (PPNAD) as part of Carney’s complex, an autosomal dominant multiple neoplasia condition associated with cardiac myxomas, hyperlentiginosis, Sertoli cell tumors, and PPNAD. PPNAD can present as micronodular or macronodular hyperplasia, or both. Phosphodiesterases can influence intracellular cAMP and can thereby impact PKA activation. Mutations in PDE11A and PDE8B have been identified in patients with bilateral adrenal hyperplasia and Cushing’s, with and without evidence of PPNAD.
Another rare cause of ACTH-independent Cushing’s is McCune-Albright syndrome, also associated with polyostotic fibrous dysplasia, unilateral café-au-lait spots, and precocious puberty. McCune-Albright syndrome is caused by activating mutations in the stimulatory G protein alpha subunit 1, GNAS-1 (guanine nucleotide binding protein alpha stimulating activity polypeptide 1), and such mutations have also been found in bilateral macronodular hyperplasia without other McCune-Albright features and, in rare instances, also in isolated cortisol-producing adrenal adenomas (Table 406-1; Chap. 426e).
Clinical Manifestations Glucocorticoids affect almost all cells of the body, and thus signs of cortisol excess impact multiple physiologic systems (Table 406-2), with upregulation of gluconeogenesis, lipolysis, and protein catabolism causing the most prominent features. In addition, excess glucocorticoid secretion overcomes the ability of 11β-HSD2 to rapidly inactivate cortisol to cortisone in the kidney, thereby exerting mineralocorticoid actions, manifest as diastolic hypertension, hypokalemia, and edema. Excess glucocorticoids also interfere with central regulatory systems, leading to suppression of gonadotropins with subsequent hypogonadism and amenorrhea, and suppression of the hypothalamic-pituitary-thyroid axis, resulting in decreased thyroid-stimulating hormone (TSH) secretion.
SIGNS AND SYMPTOMS OF CUSHING’S SYNDROME |
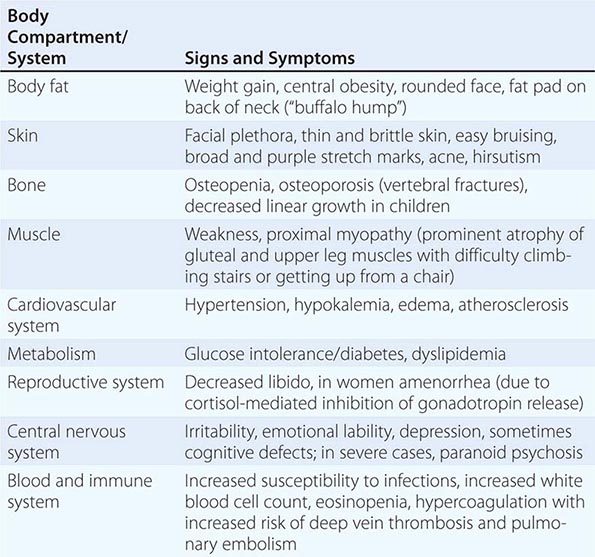
The majority of clinical signs and symptoms observed in Cushing’s syndrome are relatively nonspecific and include features such as obesity, diabetes, diastolic hypertension, hirsutism, and depression that are commonly found in patients who do not have Cushing’s. Therefore, careful clinical assessment is an important aspect of evaluating suspected cases. A diagnosis of Cushing’s should be considered when several clinical features are found in the same patient, in particular when more specific features are found. These include fragility of the skin, with easy bruising and broad (>1 cm), purplish striae (Fig. 406-9), and signs of proximal myopathy, which becomes most obvious when trying to stand up from a chair without the use of hands or when climbing stairs. Clinical manifestations of Cushing’s do not differ substantially among the different causes of Cushing’s. In ectopic ACTH syndrome, hyperpigmentation of the knuckles, scars, or skin areas exposed to increased friction can be observed (Fig. 406-9) and is caused by stimulatory effects of excess ACTH and other POMC cleavage products on melanocyte pigment production. Furthermore, patients with ectopic ACTH syndrome, and some with adrenocortical carcinoma as the cause of Cushing’s, may have a more brisk onset and rapid progression of clinical signs and symptoms.
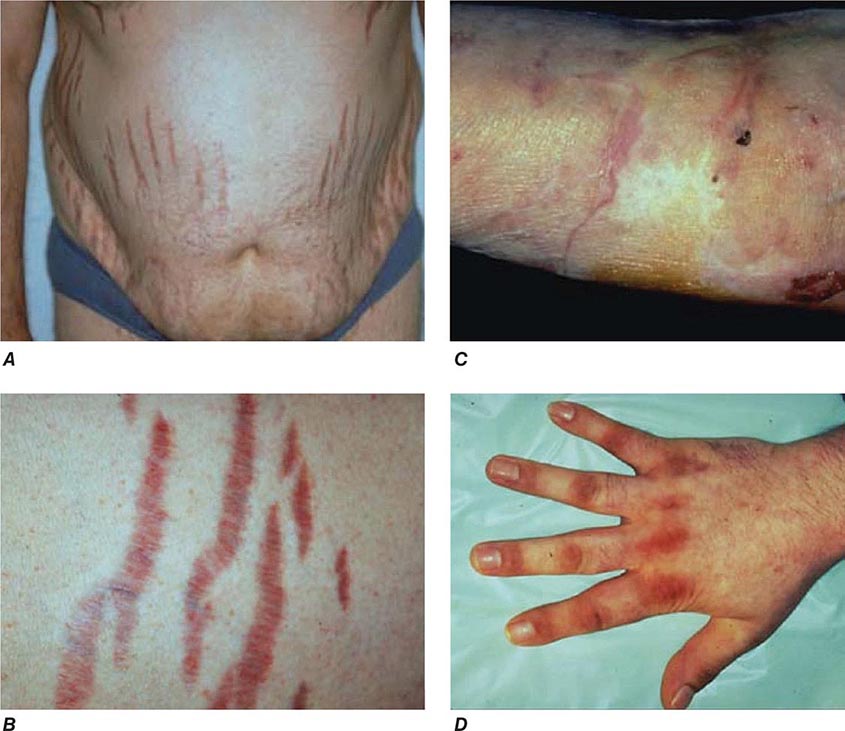
FIGURE 406-9 Clinical features of Cushing’s syndrome. A. Note central obesity and broad, purple stretch marks (B. close-up). C. Note thin and brittle skin in an elderly patient with Cushing’s syndrome. D. Hyperpigmentation of the knuckles in a patient with ectopic adrenocorticotropic hormone (ACTH) excess.
Patients with Cushing’s syndrome can be acutely endangered by deep vein thrombosis, with subsequent pulmonary embolism due to a hypercoagulable state associated with Cushing’s. The majority of patients also experience psychiatric symptoms, mostly in the form of anxiety or depression, but acute paranoid or depressive psychosis may also occur. Even after cure, long-term health may be affected by persistently impaired health-related quality of life and increased risk of cardiovascular disease and osteoporosis with vertebral fractures, depending on the duration and degree of exposure to significant cortisol excess.
Diagnosis The most important first step in the management of patients with suspected Cushing’s syndrome is to establish the correct diagnosis. Most mistakes in clinical management, leading to unnecessary imaging or surgery, are made because the diagnostic protocol is not followed (Fig. 406-10). This protocol requires establishing the diagnosis of Cushing’s beyond doubt prior to employing any tests used for the differential diagnosis of the condition. In principle, after excluding exogenous glucocorticoid use as the cause of clinical signs and symptoms, suspected cases should be tested if there are multiple and progressive features of Cushing’s, particularly features with a potentially higher discriminatory value. Exclusion of Cushing’s is also indicated in patients with incidentally discovered adrenal masses.
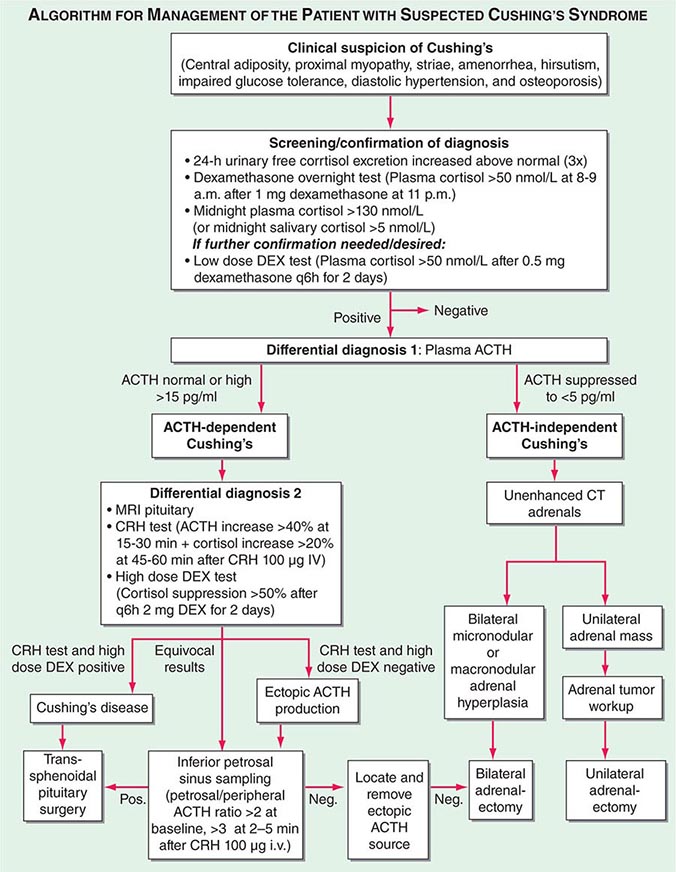
FIGURE 406-10 Management of the patient with suspected Cushing’s syndrome. ACTH, adrenocorticotropic hormone; CRH, corticotropin-releasing hormone; CT, computed tomography; DEX, dexamethasone; MRI, magnetic resonance imaging.
A diagnosis of Cushing’s can be considered as established if the results of several tests are consistently suggestive of Cushing’s. These tests may include increased 24-h urinary free cortisol excretion in three separate collections, failure to appropriately suppress morning cortisol after overnight exposure to dexamethasone, and evidence of loss of diurnal cortisol secretion with high levels at midnight, the time of the physiologically lowest secretion (Fig. 406-10). Factors potentially affecting the outcome of these diagnostic tests have to be excluded such as incomplete 24-h urine collection or rapid inactivation of dexamethasone due to concurrent intake of CYP3A4-inducing drugs (e.g., antiepileptics, rifampicin). Concurrent intake of oral contraceptives that raise CBG and thus total cortisol can cause failure to suppress after dexamethasone. If in doubt, testing should be repeated after 4–6 weeks off estrogens. Patients with pseudo-Cushing states, i.e., alcohol-related, and those with cyclic Cushing’s may require further testing to safely confirm or exclude the diagnosis of Cushing’s. In addition, the biochemical assays employed can affect the test results, with specificity representing a common problem with antibody-based assays for the measurement of urinary free cortisol. These assays have been greatly improved by the introduction of highly specific tandem mass spectrometry.
Differential Diagnosis The evaluation of patients with confirmed Cushing’s should be carried out by an endocrinologist and begins with the differential diagnosis of ACTH-dependent and ACTH-independent cortisol excess (Fig. 406-10). Generally, plasma ACTH levels are suppressed in cases of autonomous adrenal cortisol excess, as a consequence of enhanced negative feedback to the hypothalamus and pituitary. By contrast, patients with ACTH-dependent Cushing’s have normal or increased plasma ACTH, with very high levels being found in some patients with ectopic ACTH syndrome. Importantly, imaging should only be used after it is established whether the cortisol excess is ACTH-dependent or ACTH-independent, because nodules in the pituitary or the adrenal are a common finding in the general population. In patients with confirmed ACTH-independent excess, adrenal imaging is indicated (Fig. 406-11), preferably using an unenhanced computed tomography (CT) scan. This allows assessment of adrenal morphology and determination of precontrast tumor density in Hounsfield units (HU), which helps to distinguish between benign and malignant adrenal lesions.
FIGURE 406-11 Adrenal imaging in Cushing’s syndrome. A. Adrenal computed tomography (CT) showing normal bilateral adrenal morphology (arrows). B. CT scan depicting a right adrenocortical adenoma (arrow) causing Cushing’s syndrome. C. Magnetic resonance imaging (MRI) showing bilateral adrenal hyperplasia due to excess adrenocorticotropic hormone stimulation in Cushing’s disease. D. MRI showing bilateral macronodular hyperplasia causing Cushing’s syndrome.
For ACTH-dependent cortisol excess (Chap. 403), a magnetic resonance image (MRI) of the pituitary is the investigation of choice, but it may not show an abnormality in up to 40% of cases because of small tumors below the sensitivity of detection. Characteristically, pituitary corticotrope adenomas fail to enhance following gadolinium administration on T1-weighted MRI images. In all cases of confirmed ACTH-dependent Cushing’s, further tests are required for the differential diagnosis of pituitary Cushing’s disease and ectopic ACTH syndrome. These tests exploit the fact that most pituitary corticotrope adenomas still display regulatory features, including residual ACTH suppression by high-dose glucocorticoids and CRH responsiveness. In contrast, ectopic sources of ACTH are typically resistant to dexamethasone suppression and unresponsive to CRH (Fig. 406-10). However, it should be noted that a small minority of ectopic ACTH-producing tumors exhibit dynamic responses similar to pituitary corticotrope tumors. If the two tests show discordant results, or if there is any other reason for doubt, the differential diagnosis can be further clarified by performing bilateral inferior petrosal sinus sampling (IPSS) with concurrent blood sampling for ACTH in the right and left inferior petrosal sinus and a peripheral vein. An increased central/peripheral plasma ACTH ratio >2 at baseline and >3 at 2–5 min after CRH injection is indicative of Cushing’s disease (Fig. 406-10), with very high sensitivity and specificity. Of note, the results of the IPSS cannot be reliably used for lateralization (i.e., prediction of the location of the tumor within the pituitary), because there is broad interindividual variability in the venous drainage of the pituitary region. Importantly, no cortisol-lowering agents should be used prior to IPSS.
If the differential diagnostic testing indicates ectopic ACTH syndrome, then further imaging should include high-resolution, fine-cut CT scanning of the chest and abdomen for scrutiny of the lung, thymus, and pancreas. If no lesions are identified, an MRI of the chest can be considered because carcinoid tumors usually show high signal intensity on T2-weighted images. Furthermore, octreotide scintigraphy can be helpful in some cases because ectopic ACTH-producing tumors often express somatostatin receptors. Depending on the suspected cause, patients with ectopic ACTH syndrome should also undergo blood sampling for fasting gut hormones, chromogranin A, calcitonin, and biochemical exclusion of pheochromocytoma.
CUSHING’S SYNDROME |
Overt Cushing’s is associated with a poor prognosis if left untreated. In ACTH-independent disease, treatment consists of surgical removal of the adrenal tumor. For smaller tumors, a minimally invasive approach can be used, whereas for larger tumors and those suspected of malignancy, an open approach is preferred.
In Cushing’s disease, the treatment of choice is selective removal of the pituitary corticotrope tumor, usually via an endoscopic transsphenoidal approach. This results in an initial cure rate of 70–80% when performed by a highly experienced surgeon. However, even after initial remission following surgery, long-term follow-up is important because late relapse occurs in a significant number of patients. If pituitary disease recurs, there are several options, including second surgery, radiotherapy, stereotactic radiosurgery, and bilateral adrenalectomy. These options need to be applied in a highly individualized fashion.
In some patients with very severe, overt Cushing’s (e.g., difficult to control hypokalemic hypertension or acute psychosis), it may be necessary to introduce medical therapy to rapidly control the cortisol excess during the period leading up to surgery. Similarly, patients with metastasized, glucocorticoid-producing carcinomas may require long-term antiglucocorticoid drug treatment. In case of ectopic ACTH syndrome, in which the tumor cannot be located, one must carefully weigh whether drug treatment or bilateral adrenalectomy is the most appropriate choice, with the latter facilitating immediate cure but requiring life-long corticosteroid replacement. In this instance, it is paramount to ensure regular imaging follow-up for identification of the ectopic ACTH source.
Oral agents with established efficacy in Cushing’s syndrome are metyrapone and ketoconazole. Metyrapone inhibits cortisol synthesis at the level of 11β-hydroxylase (Fig. 406-1), whereas the antimycotic drug ketoconazole inhibits the early steps of steroidogenesis. Typical starting doses are 500 mg tid for metyrapone (maximum dose, 6 g) and 200 mg tid for ketoconazole (maximum dose, 1200 mg). Mitotane, a derivative of the insecticide o,p’DDD, is an adrenolytic agent that is also effective for reducing cortisol. Because of its side effect profile, it is most commonly used in the context of adrenocortical carcinoma, but low-dose treatment (500–1000 mg/d) has also been used in benign Cushing’s. In severe cases of cortisol excess, etomidate can be used to lower cortisol. It is administered by continuous IV infusion in low, nonanesthetic doses.
After the successful removal of an ACTH- or cortisol-producing tumor, the HPA axis will remain suppressed. Thus, hydrocortisone replacement needs to be initiated at the time of surgery and slowly tapered following recovery, to allow physiologic adaptation to normal cortisol levels. Depending on degree and duration of cortisol excess, the HPA axis may require many months or even years to resume normal function.
MINERALOCORTICOID EXCESS
Epidemiology Following the first description of a patient with an aldosterone-producing adrenal adenoma (Conn’s syndrome), mineralocorticoid excess was thought to represent a rare cause of hypertension. However, in studies systematically screening all patients with hypertension, a much higher prevalence is now recognized, ranging from 5 to 12%. The prevalence is higher when patients are preselected for hypokalemic hypertension.
Etiology The most common cause of mineralocorticoid excess is primary aldosteronism, reflecting excess production of aldosterone by the adrenal zona glomerulosa. Bilateral micronodular hyperplasia is somewhat more common than unilateral adrenal adenomas (Table 406-3). Somatic mutations in channels and enzymes responsible for increasing sodium and calcium influx in adrenal zona glomerulosa cells have been identified as prevalent causes of aldosterone-producing adrenal adenomas (Table 406-3) and, in the case of germline mutations, also of primary aldosteronism due to bilateral macronodular adrenal hyperplasia. However, bilateral adrenal hyperplasia as a cause of mineralocorticoid excess is usually micronodular but can also contain larger nodules that might be mistaken for a unilateral adenoma. In rare instances, primary aldosteronism is caused by an adrenocortical carcinoma. Carcinomas should be considered in younger patients and in those with larger tumors, because benign aldosterone-producing adenomas usually measure <2 cm in diameter.
CAUSES OF MINERALOCORTICOID EXCESS |
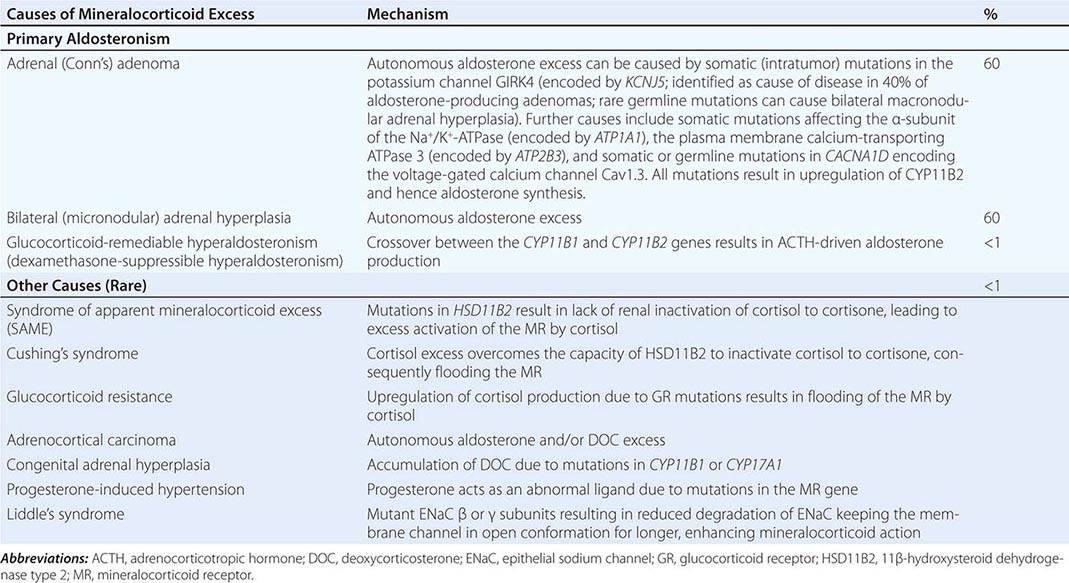
A rare cause of aldosterone excess is glucocorticoid-remediable aldosteronism (GRA), which is caused by a chimeric gene resulting from cross-over of promoter sequences between the CYP11B1 and CYP11B2 genes that are involved in glucocorticoid and mineralocorticoid synthesis, respectively (Fig. 406-1). This rearrangement brings CYP11B2 transcription under the control of ACTH receptor signaling; consequently, aldosterone production is regulated by ACTH rather than by renin. The family history can be helpful because there may be evidence for dominant transmission of hypertension. Recognition of the disorder is important because it can be associated with early-onset hypertension and strokes. In addition, glucocorticoid suppression can reduce aldosterone production.
Other rare causes of mineralocorticoid excess are listed in Table 406-3. An important cause is excess binding and activation of the mineralocorticoid receptor by a steroid other than aldosterone. Cortisol acts as a potent mineralocorticoid if it escapes efficient inactivation to cortisone by 11β-HSD2 in the kidney (Fig. 406-7). This can be caused by inactivating mutations in the HSD11B2 gene resulting in the syndrome of apparent mineralocorticoid excess (SAME) that characteristically manifests with severe hypokalemic hypertension in childhood. However, milder mutations may cause normokalemic hypertension manifesting in adulthood (type II SAME). Inhibition of 11β-HSD2 by excess licorice ingestion also results in hypokalemic hypertension, as does overwhelming of 11β-HSD2 conversion capacity by cortisol excess in Cushing’s syndrome. Deoxycorticosterone (DOC) also binds and activates the mineralocorticoid receptor and can cause hypertension if its circulating concentrations are increased. This can arise through autonomous DOC secretion by an adrenocortical carcinoma, but also when DOC accumulates as a consequence of an adrenal enzymatic block, as seen in congenital adrenal hyperplasia due to CYP11B1 (11β-hydroxylase) or CYP17A1 (17α-hydroxylase) deficiency (Fig. 406-1). Progesterone can cause hypokalemic hypertension in rare individuals who harbor a mineralocorticoid receptor mutation that enhances binding and activation by progesterone; physiologically, progesterone normally exerts antimineralocorticoid activity. Finally, excess mineralocorticoid activity can be caused by mutations in the β or γ subunits of the ENaC, disrupting its interaction with Nedd4 (Fig. 406-7), and thereby decreasing receptor internalization and degradation. The constitutively active ENAC drives hypokalemic hypertension, resulting in an autosomal dominant disorder termed Liddle’s syndrome.
Clinical Manifestations Excess activation of the mineralocorticoid receptor leads to potassium depletion and increased sodium retention, with the latter causing an expansion of extracellular and plasma volume. Increased ENaC activity also results in hydrogen depletion that can cause metabolic alkalosis. Aldosterone also has direct effects on the vascular system, where it increases cardiac remodeling and decreases compliance. Aldosterone excess may cause direct damage to the myocardium and the kidney glomeruli, in addition to secondary damage due to systemic hypertension.
The clinical hallmark of mineralocorticoid excess is hypokalemic hypertension; serum sodium tends to be normal due to the concurrent fluid retention, which in some cases can lead to peripheral edema. Hypokalemia can be exacerbated by thiazide drug treatment, which leads to increased delivery of sodium to the distal renal tubule, thereby driving potassium excretion. Severe hypokalemia can be associated with muscle weakness, overt proximal myopathy, or even hypokalemic paralysis. Severe alkalosis contributes to muscle cramps and, in severe cases, can cause tetany.
Diagnosis Diagnostic screening for mineralocorticoid excess is not currently recommended for all patients with hypertension, but should be restricted to those who exhibit hypertension associated with drug resistance, hypokalemia, an adrenal mass, or onset of disease before the age of 40 years (Fig. 406-12). The accepted screening test is concurrent measurement of plasma renin and aldosterone with subsequent calculation of the aldosterone-renin ratio (ARR) (Fig. 406-12); serum potassium needs to be normalized prior to testing. Stopping antihypertensive medication can be cumbersome, particularly in patients with severe hypertension. Thus, for practical purposes, in the first instance the patient can remain on the usual antihypertensive medications, with the exception that mineralocorticoid receptor antagonists need to be ceased at least 4 weeks prior to ARR measurement. The remaining antihypertensive drugs usually do not affect the outcome of ARR testing, except that beta blocker treatment can cause false-positive results and ACE/AT1R inhibitors can cause false-negative results in milder cases (Table 406-4).
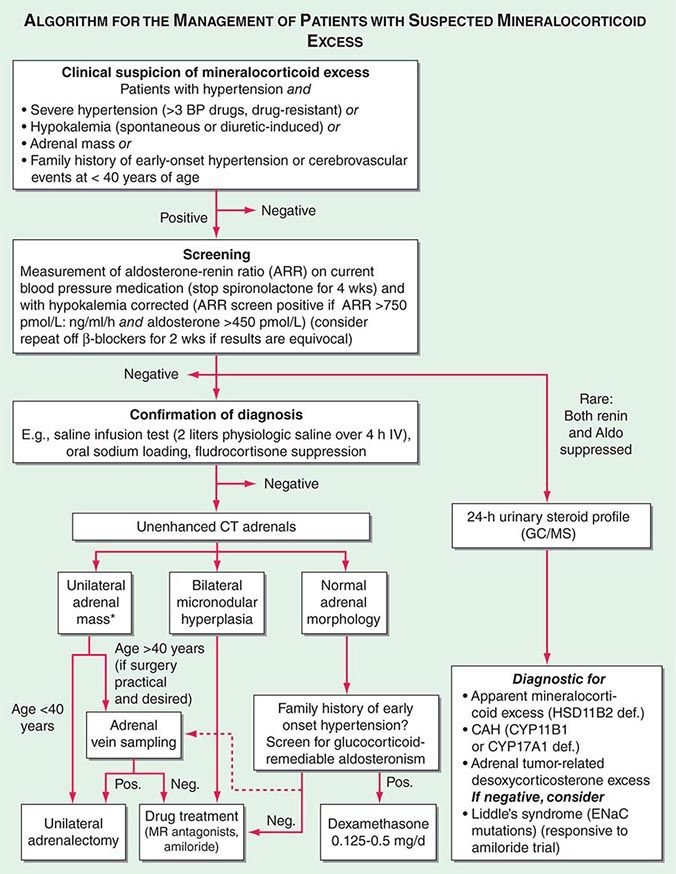
FIGURE 406-12 Management of patients with suspected mineralocorticoid excess. *Perform adrenal tumor workup (see Fig. 406-13). BP, blood pressure; CAH, congenital adrenal hyperplasia; CT, computed tomography; GC/MS, gas chromatography/mass spectrometry; PRA, plasma renin activity.
EFFECTS OF ANTIHYPERTENSIVE DRUGS ON THE ALDOSTERONE-RENIN-RATIO (ARR) |
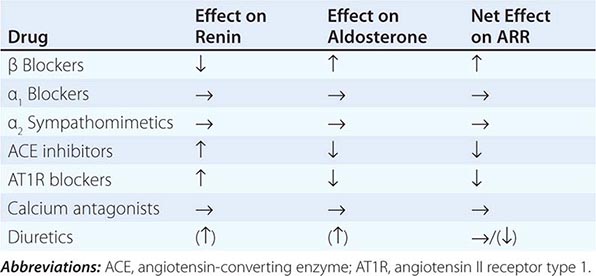
ARR screening is positive if the ratio is >750 pmol/L per ng/mL per hour, with a concurrently high normal or increased aldosterone (Fig. 406-12). If one relies on the ARR only, the likelihood of a false-positive ARR becomes greater when renin levels are very low. The characteristics of the biochemical assays are also important. Some labs measure plasma renin activity, whereas others measure plasma renin concentrations. Antibody-based assays for the measurement of serum aldosterone lack the reliability of tandem mass spectrometry assays, but these are not yet ubiquitously available.
Diagnostic confirmation of mineralocorticoid excess in a patient with positive ARR screening result should be undertaken by an endocrinologist as the tests lack optimized validation. The most straightforward is the saline infusion test, which involves the IV administration of 2 L of physiologic saline over a 4-h period. Failure of aldosterone to suppress below 140 pmol/L (5 ng/dL) is indicative of autonomous mineralocorticoid excess. Alternative tests are the oral sodium loading test (300 mmol NaCl/d for 3 days) or the fludrocortisone suppression test (0.1 mg q6h with 30 mmol NaCl q8h for 4 days); the latter can be difficult because of the risk of profound hypokalemia and increased hypertension. In patients with overt hypokalemic hypertension, strongly positive ARR, and concurrently increased aldosterone levels, confirmatory testing is usually not necessary.
Differential Diagnosis and Treatment After the diagnosis of hyperaldosteronism is established, the next step is to use adrenal imaging to further assess the cause. Fine-cut CT scanning of the adrenal region is the method of choice because it provides excellent visualization of adrenal morphology. CT will readily identify larger tumors suspicious of malignancy but may miss lesions smaller than 5 mm. The differentiation between bilateral micronodular hyperplasia and a unilateral adenoma is only required if a surgical approach is feasible and desired. Consequently, selective adrenal vein sampling (AVS) should only be carried out in surgical candidates with either no obvious lesion on CT or evidence of a unilateral lesion in patients older than 40 years, because the latter patients have a high likelihood of harboring a coincidental, endocrine-inactive adrenal adenoma (Fig. 406-12). AVS is used to compare aldosterone levels in the inferior vena cava and between the right and left adrenal veins. AVS requires concurrent measurement of cortisol to document correct placement of the catheter in the adrenal veins and should demonstrate a cortisol gradient >3 between the vena cava and each adrenal vein. Lateralization is confirmed by an aldosterone/cortisol ratio that is at least twofold higher on one side than the other. AVS is a complex procedure that requires a highly skilled interventional radiologist. Even then, the right adrenal vein can be difficult to cannulate correctly, which, if not achieved, invalidates the procedure. There is also no agreement as to whether the two adrenal veins should be cannulated simultaneously or successively and whether ACTH stimulation enhances the diagnostic value of AVS.
Patients younger than 40 years with confirmed mineralocorticoid excess and a unilateral lesion on CT can go straight to surgery, which is also indicated in patients with confirmed lateralization documented by a valid AVS procedure. Laparoscopic adrenalectomy is the preferred approach. Patients who are not surgical candidates, or with evidence of bilateral hyperplasia based on CT or AVS, should be treated medically (Fig. 406-12). Medical treatment, which can also be considered prior to surgery to avoid postsurgical hypoaldosteronism, consists primarily of the mineralocorticoid receptor antagonist spironolactone. It can be started at 12.5–50 mg bid and titrated up to a maximum of 400 mg/d to control blood pressure and normalize potassium. Side effects include menstrual irregularity, decreased libido, and gynecomastia. The more selective MR antagonist eplerenone can also be used. Doses start at 25 mg bid, and it can be titrated up to 200 mg/d. Another useful drug is the sodium channel blocker amiloride (5–10 mg bid).
In patients with normal adrenal morphology and family history of early-onset, severe hypertension, a diagnosis of GRA should be considered and can be evaluated using genetic testing. Treatment of GRA consists of administering dexamethasone, using the lowest dose possible to control blood pressure. Some patients also require additional MR antagonist treatment.
The diagnosis of nonaldosterone-related mineralocorticoid excess is based on documentation of suppressed renin and suppressed aldosterone in the presence of hypokalemic hypertension. This testing is best carried out by employing urinary steroid metabolite profiling by gas chromatography/mass spectrometry (GC/MS). An increased free cortisol over free cortisone ratio is suggestive of SAME and can be treated with dexamethasone. Steroid profiling by GC/MS also detects the steroids associated with CYP11B1 and CYP17A1 deficiency or the irregular steroid secretion pattern in a DOC-producing adrenocortical carcinoma (Fig. 406-12). If the GC/MS profile is normal, then Liddle’s syndrome should be considered. It is very sensitive to amiloride treatment but will not respond to MR antagonist treatment, because the defect is due to a constitutively active ENaC.
APPROACH TO THE PATIENT: INCIDENTALLY DISCOVERED ADRENAL MASS
Epidemiology Incidentally discovered adrenal masses, commonly termed adrenal “incidentalomas,” are common, with a prevalence of at least 2% in the general population as documented in CT and autopsy series. The prevalence increases with age, with 1% of 40-year-olds and 7% of 70-year-olds harboring an adrenal mass.
Etiology Most solitary adrenal tumors are monoclonal neoplasms. Several genetic syndromes, including MEN 1 (MEN1), MEN 2 (RET), Carney’s complex (PRKAR1A), and McCune-Albright (GNAS1), can have adrenal tumors as one of their features. Somatic mutations in MEN1, GNAS1, and PRKAR1A have been identified in a small proportion of sporadic adrenocortical adenomas. Aberrant expression of membrane receptors (gastric inhibitory peptide, α- and β-adrenergic, luteinizing hormone, vasopressin V1, and interleukin 1 receptors) have been identified in some sporadic cases of macronodular adrenocortical hyperplasia.
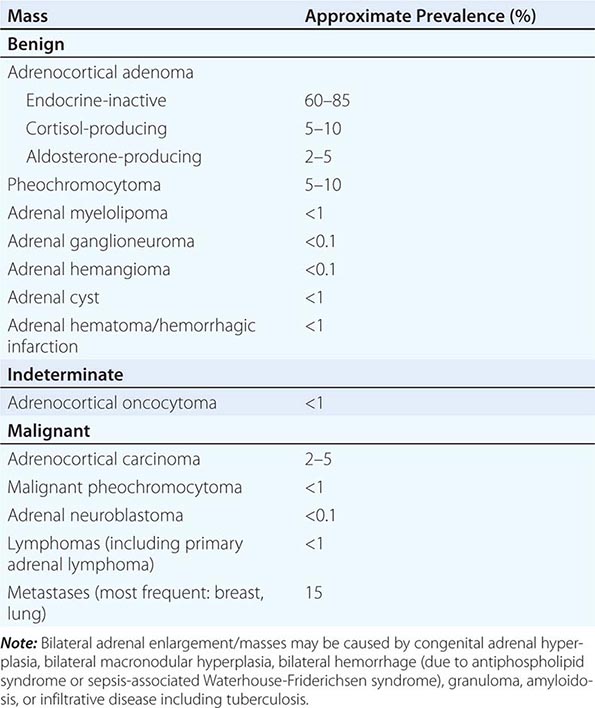
The majority of adrenal nodules are endocrine-inactive adrenocortical adenomas. However, larger series suggest that up to 25% of adrenal nodules are hormonally active, due to a cortisol- or aldosterone-producing adrenocortical adenoma or a pheochromocytoma associated with catecholamine excess (Table 406-5). Adrenocortical carcinoma is rare but is the cause of an adrenal mass in 5% of patients. However, the most common cause of a malignant adrenal mass is metastasis originating from another solid tissue tumor (Table 406-5).
CLASSIFICATION OF UNILATERAL ADRENAL MASSES |
Differential Diagnosis and Treatment Patients with an adrenal mass >1 cm require a diagnostic evaluation. Two key questions need to be addressed: (1) Does the tumor autonomously secrete hormones that could have a detrimental effect on health? (2) Is the adrenal mass benign or malignant?
Hormone secretion by an adrenal mass occurs along a continuum, with a gradual increase in clinical manifestations in parallel with hormone levels. Exclusion of catecholamine excess from a pheochromocytoma arising from the adrenal medulla is a mandatory part of the diagnostic workup (Fig. 406-13). Furthermore, autonomous cortisol and aldosterone secretion resulting in Cushing’s syndrome or primary aldosteronism, respectively, require exclusion. Adrenal incidentalomas can be associated with lower levels of autonomous cortisol secretion, and patients may lack overt clinical features of Cushing’s syndrome. Nonetheless, they may exhibit one or more components of the metabolic syndrome (e.g., obesity, type 2 diabetes, or hypertension). There is ongoing debate about the optimal treatment for these patients with mild or subclinical Cushing’s syndrome. Overproduction of adrenal androgen precursors, DHEA and its sulfate, is rare and most frequently seen in the context of adrenocortical carcinoma, as are increased levels of steroid precursors such as 17-hydroxyprogesterone.
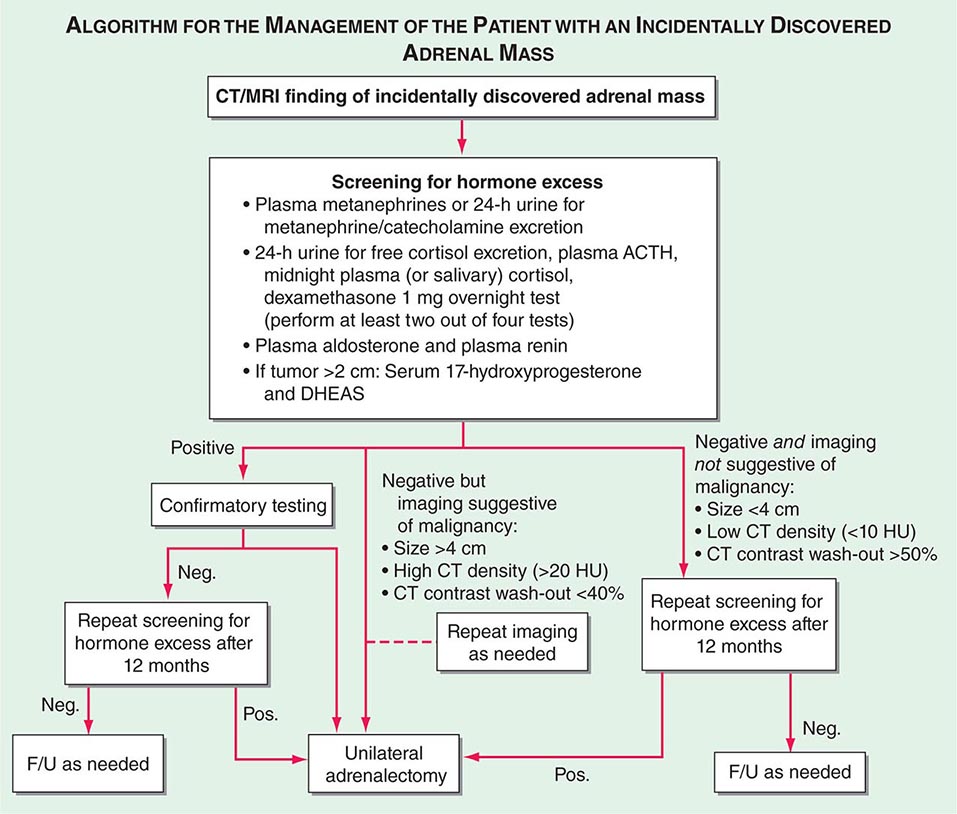
FIGURE 406-13 Management of the patient with an incidentally discovered adrenal mass. CT, computed tomography; F/U, follow-up; MRI, magnetic resonance imaging.
For the differentiation of benign from malignant adrenal masses, imaging is relatively sensitive, although specificity is suboptimal. CT is the procedure of choice for imaging the adrenal glands (Fig. 406-11). The risk of adrenocortical carcinoma, pheochromocytoma, and benign adrenal myelolipoma increases with the diameter of the adrenal mass. However, size alone is of poor predictive value, with only 80% sensitivity and 60% specificity for the differentiation of benign from malignant masses when using a 4-cm cut-off. Metastases are found with similar frequency in adrenal masses of all sizes. Tumor density on unenhanced CT is of additional diagnostic value, with most adrenocortical adenomas being lipid rich and thus presenting with low attenuation values (i.e., densities of <10 HU). By contrast, adrenocortical carcinomas, but also pheochromocytomas, usually have high attenuation values (i.e., densities >20 HU on precontrast scans). Generally, benign lesions are rounded and homogenous, whereas most malignant lesions appear lobulated and inhomogeneous. Pheochromocytoma and adrenomyelolipoma may also exhibit lobulated and inhomogeneous features. Additional information can be obtained from CT by assessment of contrast wash-out after 15 min, which is >50% in benign lesions but <40% in malignant lesions, which usually have a more extensive vascularization. MRI also allows for the visualization of the adrenal glands with somewhat lower resolution than CT. However, because it does not involve exposure to ionizing radiation, it is preferred in children, young adults, and during pregnancy. MRI has a valuable role in the characterization of indeterminate adrenal lesions using chemical shift analysis, with malignant tumors rarely showing loss of signal on opposed-phase MRI.
Fine-needle aspiration (FNA) or CT-guided biopsy of an adrenal mass is almost never indicated. FNA of a pheochromocytoma can cause a life-threatening hypertensive crisis. FNA of an adrenocortical carcinoma violates the tumor capsule and can cause needle track metastasis. FNA should only be considered in a patient with a history of nonadrenal malignancy and a newly detected adrenal mass, after careful exclusion of pheochromocytoma, and if the outcome will influence therapeutic management. It is important to recognize that in 25% of patients with a previous history of nonadrenal malignancy, a newly detected mass on CT is not a metastasis.
Adrenal masses associated with confirmed hormone excess or suspected malignancy are usually treated surgically (Fig. 406-13) or, if adrenalectomy is not feasible or desired, with medication. Preoperative exclusion of glucocorticoid excess is particularly important for the prediction of postoperative suppression of the contralateral adrenal gland, which requires glucocorticoid replacement peri- and postoperatively. If the initial decision is for observation, imaging and biochemical testing should be repeated about a year after the first assessment. However, this may be performed earlier in patients with borderline imaging or hormonal findings. There is no agreement with regard to the required long-term follow-up beyond 1 year in patients with normal biochemistry and no evidence of increased tumor size at follow-up.
ADRENOCORTICAL CARCINOMA
Adrenocortical carcinoma (ACC) is a rare malignancy with an annual incidence of 1–2 per million population. ACC is generally considered a highly malignant tumor; however, it presents with broad interindividual variability with regard to biologic characteristics and clinical behavior. Somatic mutations in the tumor-suppressor gene TP53 are found in 25% of apparently sporadic ACC. Germline TP53 mutations are the cause of the Li-Fraumeni syndrome associated with multiple solid organ cancers including ACC and are found in 25% of pediatric ACC cases; the TP53 mutation R337H is found in almost all pediatric ACC in Brazil. Other genetic changes identified in ACC include alterations in the Wnt/β-catenin pathway and in the insulin-like growth factor 2 (IGF2) cluster; IGF2 overexpression is found in 90% of ACC.
Patients with large adrenal tumors suspicious of malignancy should be managed by a multidisciplinary specialist team, including an endocrinologist, an oncologist, a surgeon, a radiologist, and a histopathologist. FNA is not indicated in suspected ACC: first, cytology and also histopathology of a core biopsy cannot differentiate between benign and malignant primary adrenal masses; second, FNA violates the tumor capsule and may even cause needle canal metastasis. Even when the entire tumor specimen is available, the histopathologic differentiation between benign and malignant lesions is a diagnostic challenge. The most common histopathologic classification is the Weiss score, taking into account high nuclear grade; mitotic rate (>5/HPF); atypical mitosis; <25% clear cells; diffuse architecture; and presence of necrosis, venous invasion, and invasion of sinusoidal structures and tumor capsule. The presence of three or more elements suggests ACC.
Although 60–70% of ACCs show biochemical evidence of steroid overproduction, in many patients, this is not clinically apparent due to the relatively inefficient steroid production by the adrenocortical cancer cells. Excess production of glucocorticoids and adrenal androgen precursors are most common. Mixed excess production of several corticosteroid classes by an adrenal tumor is generally indicative of malignancy.
Tumor staging at diagnosis (Table 406-6) has important prognostic implications and requires scanning of the chest and abdomen for local organ invasion, lymphadenopathy, and metastases. Intravenous contrast medium is necessary for maximum sensitivity for hepatic metastases. An adrenal origin may be difficult to determine on standard axial CT imaging if the tumors are large and invasive, but CT reconstructions and MRI are more informative (Fig. 406-14) using multiple planes and different sequences. Vascular and adjacent organ invasion is diagnostic of malignancy. 18-Fluoro-2-deoxy-D-glucose positron emission tomography (18-FDG PET) is highly sensitive for the detection of malignancy and can be used to detect small metastases or local recurrence that may not be obvious on CT (Fig. 406-14). However, FDG PET is not specific and therefore cannot be used for differentiating benign from malignant adrenal lesions. Metastasis in ACC most frequently occurs to liver and lung.
CLASSIFICATION SYSTEM FOR STAGING OF ADRENOCORTICAL CARCINOMA |
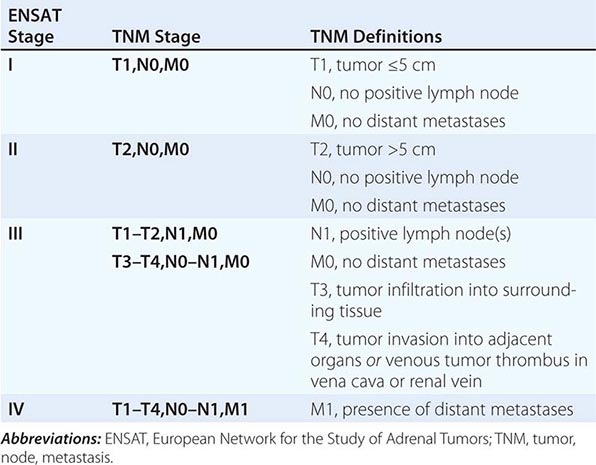
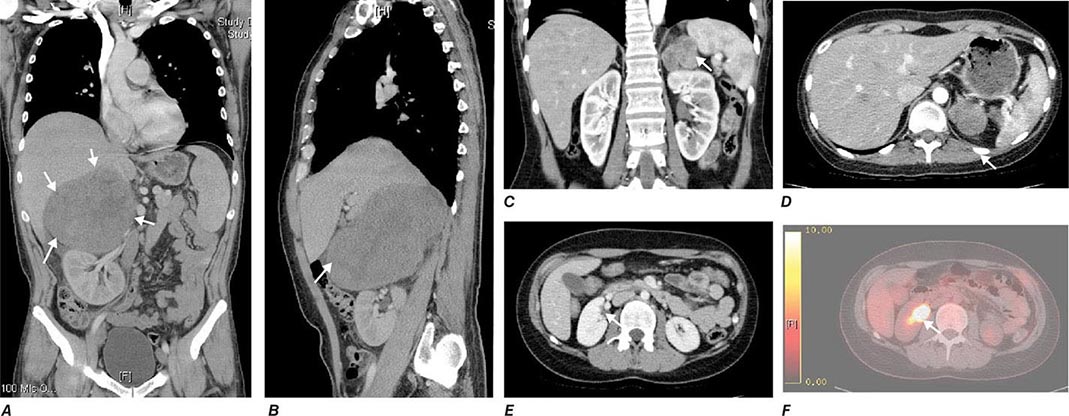
FIGURE 406-14 Imaging in adrenocortical carcinoma. Magnetic resonance imaging scan with (A) frontal and (B) lateral views of a right adrenocortical carcinoma that was detected incidentally. Computed tomography (CT) scan with (C) coronal and (D) transverse views depicting a right-sided adrenocortical carcinoma. Note the irregular border and inhomogeneous structure. CT scan (E) and positron emission tomography/CT (F) visualizing a peritoneal metastasis of an adrenocortical carcinoma in close proximity to the right kidney (arrow).
There is no established grading system for ACC, and the Weiss score carries no prognostic value; the most important prognostic histopathologic parameter is the Ki67 proliferation index, with Ki67 <10% indicative of slow to moderate growth velocity, whereas a Ki67 ≥10% is associated with poor prognosis including high risk of recurrence and rapid progression.
Cure of ACC can only be achieved by early detection and complete surgical removal. Capsule violation during primary surgery, metastasis at diagnosis, and primary treatment in a nonspecialist center are major determinants of poor survival. If the primary tumor invades adjacent organs, en bloc removal of kidney and spleen should be considered to reduce the risk of recurrence. Surgery can also be considered in a patient with metastases if there is severe tumor-related hormone excess. This indication needs to be carefully weighed against surgical risk, including thromboembolic complications, and the resulting delay in the introduction of other therapeutic options. Patients with confirmed ACC and successful removal of the primary tumor should receive adjuvant treatment with mitotane (o,p’DDD), particularly in patients with a high risk of recurrence as determined by tumor size >8 cm, histopathologic signs of vascular invasion, capsule invasion or violation, and a Ki67 proliferation index ≥10%. Adjuvant mitotane should be continued for at least 2 years, if the patient can tolerate side effects. Regular monitoring of plasma mitotane levels is mandatory (therapeutic range 14–20 mg/L; neurotoxic complications more frequent at >20 mg/L). Mitotane is usually started at 500 mg tid, with stepwise increases to a maximum dose of 2000 mg tid in days (high-dose saturation) or weeks (low-dose saturation) as tolerated. Once therapeutic range plasma mitotane levels are achieved, the dose can be tapered to maintenance doses mostly ranging from 1000 to 1500 mg tid. Mitotane treatment results in disruption of cortisol synthesis and thus requires glucocorticoid replacement; glucocorticoid replacement dose should be at least double of that usually used in adrenal insufficiency (i.e., 20 mg tid) because mitotane induces hepatic CYP3A4 activity resulting in rapid inactivation of glucocorticoids. Mitotane also increases circulating CBG, thereby decreasing the available free cortisol fraction. Single metastases can be addressed surgically or with radiofrequency ablation as appropriate. If the tumor recurs or progresses during mitotane treatment, chemotherapy should be considered; the established first-line chemotherapy regimen is the combination of cisplatin, etoposide, and doxorubicin plus continuing mitotane. Painful bone metastasis responds to irradiation. Overall survival in ACC is still poor, with 5-year survival rates of 30–40% and a median survival of 15 months in metastatic ACC.
ADRENAL INSUFFICIENCY
Epidemiology The prevalence of well-documented, permanent adrenal insufficiency is 5 in 10,000 in the general population. Hypothalamic-pituitary origin of disease is most frequent, with a prevalence of 3 in 10,000, whereas primary adrenal insufficiency has a prevalence of 2 in 10,000. Approximately one-half of the latter cases are acquired, mostly caused by autoimmune destruction of the adrenal glands; the other one-half are genetic, most commonly caused by distinct enzymatic blocks in adrenal steroidogenesis affecting glucocorticoid synthesis (i.e., congenital adrenal hyperplasia.)
Adrenal insufficiency arising from suppression of the HPA axis as a consequence of exogenous glucocorticoid treatment is much more common, occurring in 0.5–2% of the population in developed countries.
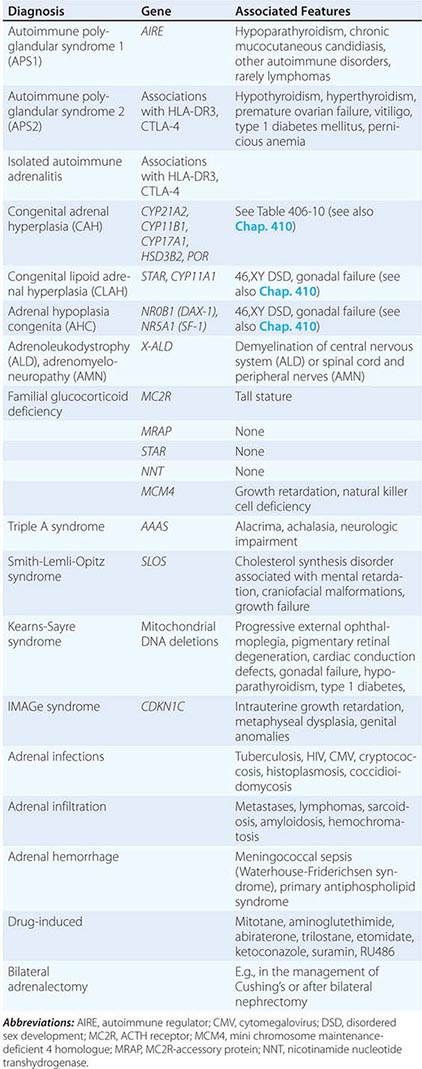
Etiology Primary adrenal insufficiency is most commonly caused by autoimmune adrenalitis. Isolated autoimmune adrenalitis accounts for 30–40%, whereas 60–70% develop adrenal insufficiency as part of autoimmune polyglandular syndromes (APS) (Chap. 408) (Table 406-7). APS1, also termed APECED (autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy), is the underlying cause in 10% of patients affected by APS. APS1 is transmitted in an autosomal recessive manner and is caused by mutations in the autoimmune regulator gene AIRE. Associated autoimmune conditions overlap with those seen in APS2, but may also include total alopecia, primary hypoparathyroidism, and, in rare cases, lymphoma. APS1 patients invariably develop chronic mucocutaneous candidiasis, usually manifest in childhood, and preceding adrenal insufficiency by years or decades. The much more prevalent APS2 is of polygenic inheritance, with confirmed associations with the HLA-DR3 gene region in the major histocompatibility complex and distinct gene regions involved in immune regulation (CTLA-4, PTPN22, CLEC16A). Coincident autoimmune disease most frequently includes thyroid autoimmune disease, vitiligo, and premature ovarian failure. Less commonly, additional features may include type 1 diabetes and pernicious anemia caused by vitamin B12 deficiency.
CAUSES OF PRIMARY ADRENAL INSUFFICIENCY |
X-linked adrenoleukodystrophy has an incidence of 1:20,000 males and is caused by mutations in the X-ALD gene encoding the peroxisomal membrane transporter protein ABCD1; its disruption results in accumulation of very long chain (>24 carbon atoms) fatty acids. Approximately 50% of cases manifest in early childhood with rapidly progressive white matter disease (cerebral ALD); 35% present during adolescence or in early adulthood with neurologic features indicative of myelin and peripheral nervous system involvement (adrenomyeloneuropathy [AMN]). In the remaining 15%, adrenal insufficiency is the sole manifestation of disease. Of note, distinct mutations manifest with variable penetrance and phenotypes within affected families.
Rarer causes of adrenal insufficiency involve destruction of the adrenal glands as a consequence of infection, hemorrhage, or infiltration (Table 406-7); tuberculous adrenalitis is still a frequent cause of disease in developing countries. Adrenal metastases rarely cause adrenal insufficiency, and this occurs only with bilateral, bulky metastases.
Inborn causes of primary adrenal insufficiency other than congenital adrenal hyperplasia are rare, causing less than 1% of cases. However, their elucidation provides important insights into adrenal gland development and physiology. Mutations causing primary adrenal insufficiency (Table 406-7) include factors regulating adrenal development and steroidogenesis (DAX-1, SF-1), cholesterol synthesis, import and cleavage (DHCR7, StAR, CYP11A1), and elements of the adrenal ACTH response pathway (MC2R, MRAP) (Fig. 406-5), and factors involved in redox regulation (NNT) and DNA repair (MCM4, CDKN1C).
Secondary adrenal insufficiency is the consequence of dysfunction of the hypothalamic-pituitary component of the HPA axis (Table 406-8). Excluding iatrogenic suppression, the overwhelming majority of cases are caused by pituitary or hypothalamic tumors or their treatment by surgery or irradiation (Chap. 403). Rarer causes include pituitary apoplexy, either as a consequence of an infarcted pituitary adenoma or transient reduction in the blood supply of the pituitary during surgery or after rapid blood loss associated with parturition, also termed Sheehan’s syndrome. Isolated ACTH deficiency is rarely caused by autoimmune disease or pituitary infiltration (Table 406-8). Mutations in the ACTH precursor POMC or in factors regulating pituitary development are genetic causes of ACTH deficiency (Table 406-8).
CAUSES OF SECONDARY ADRENAL INSUFFICIENCY |
Clinical Manifestations In principle, the clinical features of primary adrenal insufficiency (Addison’s disease) are characterized by the loss of both glucocorticoid and mineralocorticoid secretion (Table 406-9). In secondary adrenal insufficiency, only glucocorticoid deficiency is present, as the adrenal itself is intact and thus still amenable to regulation by the RAA system. Adrenal androgen secretion is disrupted in both primary and secondary adrenal insufficiency (Table 406-9). Hypothalamic-pituitary disease can lead to additional clinical manifestations due to involvement of other endocrine axes (thyroid, gonads, growth hormone, prolactin) or visual impairment with bitemporal hemianopia caused by chiasmal compression. It is important to recognize that iatrogenic adrenal insufficiency caused by exogenous glucocorticoid suppression of the HPA axis may result in all symptoms associated with glucocorticoid deficiency (Table 406-9), if exogenous glucocorticoids are stopped abruptly. However, patients will appear clinically cushingoid as a result of the preceding overexposure to glucocorticoids.
SIGNS AND SYMPTOMS OF ADRENAL INSUFFICIENCY |
Abbreviations: AVP, arginine vasopressin; TSH, thyroid-stimulating hormone.
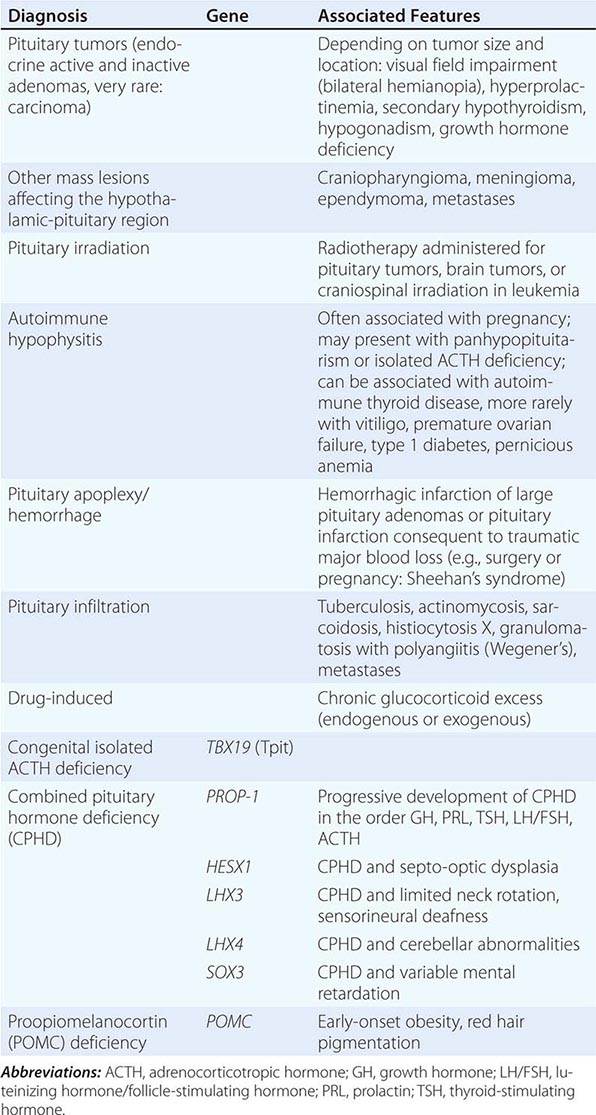
Chronic adrenal insufficiency manifests with relatively nonspecific signs and symptoms such as fatigue and loss of energy, often resulting in delayed or missed diagnoses (e.g., as depression or anorexia). A distinguishing feature of primary adrenal insufficiency is hyperpigmentation, which is caused by excess ACTH stimulation of melanocytes. Hyperpigmentation is most pronounced in skin areas exposed to increased friction or shear stress and is increased by sunlight (Fig. 406-15). Conversely, in secondary adrenal insufficiency, the skin has an alabaster-like paleness due to lack of ACTH secretion.
FIGURE 406-15 Clinical features of Addison’s disease. Note the hyperpigmentation in areas of increased friction including (A) palmar creases, (B) dorsal foot, (C) nipples and axillary region, and (D) patchy hyperpigmentation of the oral mucosa.
Hyponatremia is a characteristic biochemical feature in primary adrenal insufficiency and is found in 80% of patients at presentation. Hyperkalemia is present in 40% of patients at initial diagnosis. Hyponatremia is primarily caused by mineralocorticoid deficiency but can also occur in secondary adrenal insufficiency due to diminished inhibition of antidiuretic hormone (ADH) release by cortisol, resulting in mild syndrome of inappropriate secretion of antidiuretic hormone (SIADH). Glucocorticoid deficiency also results in slightly increased TSH concentrations that normalize within days to weeks after initiation of glucocorticoid replacement.
Acute adrenal insufficiency usually occurs after a prolonged period of nonspecific complaints and is more frequently observed in patients with primary adrenal insufficiency, due to the loss of both glucocorticoid and mineralocorticoid secretion. Postural hypotension may progress to hypovolemic shock. Adrenal insufficiency may mimic features of acute abdomen with abdominal tenderness, nausea, vomiting, and fever. In some cases, the primary presentation may resemble neurologic disease, with decreased responsiveness, progressing to stupor and coma. An adrenal crisis can be triggered by an intercurrent illness, surgical or other stress, or increased glucocorticoid inactivation (e.g., hyperthyroidism).
Diagnosis The diagnosis of adrenal insufficiency is established by the short cosyntropin test, a safe and reliable tool with excellent predictive diagnostic value (Fig. 406-16). The cut-off for failure is usually defined at cortisol levels of <500–550 nmol/L (18–20 μg/dL) sampled 30–60 min after ACTH stimulation; the exact cut-off is dependent on the locally available assay. During the early phase of HPA disruption (e.g., within 4 weeks of pituitary insufficiency), patients may still respond to exogenous ACTH stimulation. In this circumstance, the ITT is an alternative choice but is more invasive and should be carried out only under a specialist’s supervision (see above). Induction of hypoglycemia is contraindicated in individuals with diabetes mellitus, cardiovascular disease, or history of seizures. Random serum cortisol measurements are of limited diagnostic value, because baseline cortisol levels may be coincidentally low due to the physiologic diurnal rhythm of cortisol secretion (Fig. 406-3). Similarly, many patients with secondary adrenal insufficiency have relatively normal baseline cortisol levels but fail to mount an appropriate cortisol response to ACTH, which can only be revealed by stimulation testing. Importantly, tests to establish the diagnosis of adrenal insufficiency should never delay treatment. Thus, in a patient with suspected adrenal crisis, it is reasonable to draw baseline cortisol levels, provide replacement therapy, and defer formal stimulation testing until a later time.
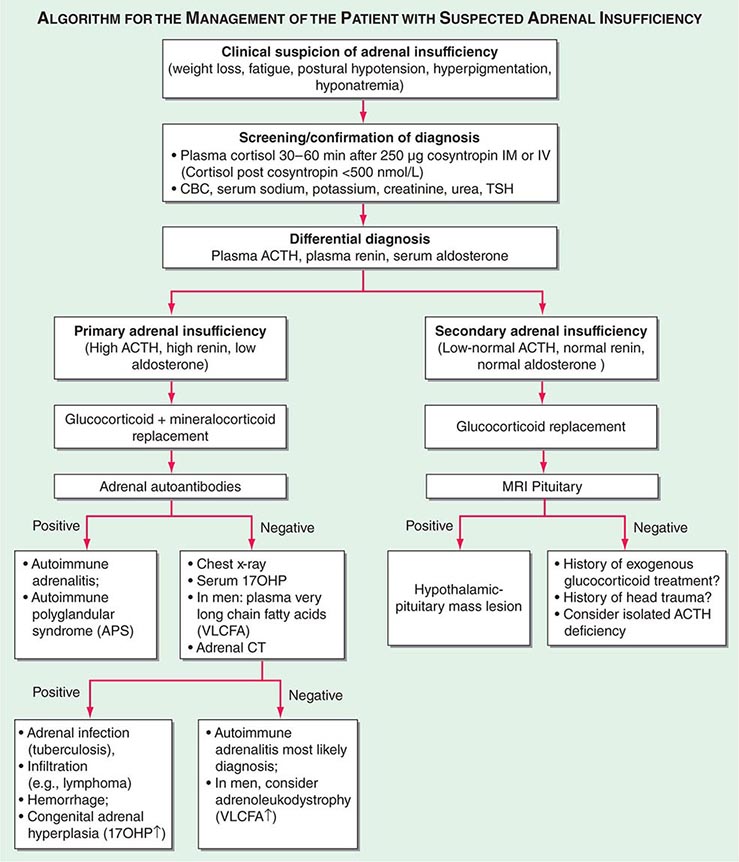
FIGURE 406-16 Management of the patient with suspected adrenal insufficiency. ACTH, adrenocorticotropic hormone; CBC, complete blood count; MRI, magnetic resonance imaging; PRA, plasma renin activity; TSH, thyroid-stimulating hormone.
Once adrenal insufficiency is confirmed, measurement of plasma ACTH is the next step, with increased or inappropriately low levels defining primary and secondary origin of disease, respectively (Fig. 406-16). In primary adrenal insufficiency, increased plasma renin will confirm the presence of mineralocorticoid deficiency. At initial presentation, patients with primary adrenal insufficiency should undergo screening for steroid autoantibodies as a marker of autoimmune adrenalitis. If these tests are negative, adrenal imaging by CT is indicated to investigate possible hemorrhage, infiltration, or masses. In male patients with negative autoantibodies in the plasma, very-long-chain fatty acids should be measured to exclude X-ALD. Patients with inappropriately low ACTH, in the presence of confirmed cortisol deficiency, should undergo hypothalamic-pituitary imaging by MRI. Features suggestive of preceding pituitary apoplexy, such as sudden-onset severe headache or history of previous head trauma, should be carefully explored, particularly in patients with no obvious MRI lesion.
TREATMENT | ACUTE ADRENAL INSUFFICIENCY |
Acute adrenal insufficiency requires immediate initiation of rehydration, usually carried out by saline infusion at initial rates of 1 L/h with continuous cardiac monitoring. Glucocorticoid replacement should be initiated by bolus injection of 100 mg hydrocortisone, followed by the administration of 100–200 mg hydrocortisone over 24 h, either by continuous infusion or by bolus IV or IM injections. Mineralocorticoid replacement can be initiated once the daily hydrocortisone dose has been reduced to <50 mg because at higher doses hydrocortisone provides sufficient stimulation of mineralocorticoid receptors.
Glucocorticoid replacement for the treatment of chronic adrenal insufficiency should be administered at a dose that replaces the physiologic daily cortisol production, which is usually achieved by the oral administration of 15–25 mg hydrocortisone in two to three divided doses. Pregnancy may require an increase in hydrocortisone dose by 50% during the last trimester. In all patients, at least one-half of the daily dose should be administered in the morning. Currently available glucocorticoid preparations fail to mimic the physiologic cortisol secretion rhythm (Fig. 406-3). Long-acting glucocorticoids such as prednisolone or dexamethasone are not preferred because they result in increased glucocorticoid exposure due to extended glucocorticoid receptor activation at times of physiologically low cortisol secretion. There are no well-established dose equivalencies, but as a guide, equipotency can be assumed for 1 mg hydrocortisone, 1.6 mg cortisone acetate, 0.2 mg prednisolone, 0.25 mg prednisone, and 0.025 mg dexamethasone.
Monitoring of glucocorticoid replacement is mainly based on the history and examination for signs and symptoms suggestive of glucocorticoid over- or underreplacement, including assessment of body weight and blood pressure. Plasma ACTH, 24-h urinary free cortisol, or serum cortisol day curves reflect whether hydrocortisone has been taken or not, but do not convey reliable information about replacement quality. In patients with isolated primary adrenal insufficiency, monitoring should include screening for autoimmune thyroid disease, and female patients should be made aware of the possibility of premature ovarian failure. Supraphysiologic glucocorticoid treatment with doses equivalent to 30 mg hydrocortisone or more will affect bone metabolism, and these patients should undergo regular bone mineral density evaluation. All patients with adrenal insufficiency need to be instructed about the requirement for stress-related glucocorticoid dose adjustments. These generally consist of doubling the routine oral glucocorticoid dose in the case of intercurrent illness with fever and bed rest and the need for IV hydrocortisone injection at a daily dose of 100 mg in cases of prolonged vomiting, surgery, or trauma. Patients living or traveling in regions with delayed access to acute health care should carry a hydrocortisone self-injection emergency kit, in addition to their usual steroid emergency cards and bracelets.
Mineralocorticoid replacement in primary adrenal insufficiency should be initiated at a dose of 100–150 μg fludrocortisone. The adequacy of treatment can be evaluated by measuring blood pressure, sitting and standing, to detect a postural drop indicative of hypovolemia. In addition, serum sodium, potassium, and plasma renin should be measured regularly. Renin levels should be kept in the upper normal reference range. Changes in glucocorticoid dose may also impact on mineralocorticoid replacement as cortisol also binds the mineralocorticoid receptor; 40 mg hydrocortisone is equivalent to 100 μg fludrocortisone. In patients living or traveling in areas with hot or tropical weather conditions, the fludrocortisone dose should be increased by 50–100 μg during the summer. Mineralocorticoid dose may also need to be adjusted during pregnancy, due to the antimineralocorticoid activity of progesterone, but this is less often required than hydrocortisone dose adjustment. Plasma renin cannot serve as a monitoring tool during pregnancy, because renin rises physiologically during gestation.
Adrenal androgen replacement is an option in patients with lack of energy, despite optimized glucocorticoid and mineralocorticoid replacement. It may also be indicated in women with features of androgen deficiency, including loss of libido. Adrenal androgen replacement can be achieved by once-daily administration of 25–50 mg DHEA. Treatment is monitored by measurement of DHEAS, androstenedione, testosterone, and sex hormone–binding globulin (SHBG) 24 h after the last DHEA dose.
CONGENITAL ADRENAL HYPERPLASIA
(See also Chap. 410) Congenital adrenal hyperplasia (CAH) is caused by mutations in genes encoding steroidogenic enzymes involved in glucocorticoid synthesis (CYP21A2, CYP17A1, HSD3B2, CYP11B1) or in the cofactor enzyme P450 oxidoreductase that serves as an electron donor to CYP21A2 and CYP17A1 (Fig. 406-1). Invariably, patients affected by CAH exhibit glucocorticoid deficiency. Depending on the exact step of enzymatic block, they may also have excess production of mineralocorticoids or deficient production of sex steroids (Table 406-10). The diagnosis of CAH is readily established by measurement of the steroids accumulating before the distinct enzymatic block, either in serum or in urine, preferably by the use of mass spectrometry–based assays (Table 406-10).
VARIANTS OF CONGENITAL ADRENAL HYPERPLASIA |

Mutations in CYP21A2 are the most prevalent cause of CAH, responsible for 90–95% of cases. 21-Hydroxylase deficiency disrupts glucocorticoid and mineralocorticoid synthesis (Fig. 406-1), resulting in diminished negative feedback via the HPA axis. This leads to increased pituitary ACTH release, which drives increased synthesis of adrenal androgen precursors and subsequent androgen excess. The degree of impairment of glucocorticoid and mineralocorticoid secretion depends on the severity of mutations. Major loss-of-function mutations result in combined glucocorticoid and mineralocorticoid deficiency (classic CAH, neonatal presentation), whereas less severe mutations affect glucocorticoid synthesis only (simple virilizing CAH, neonatal or early childhood presentation). The mildest mutations result in the least severe clinical phenotype, nonclassic CAH, usually presenting during adolescence and early adulthood and with preserved glucocorticoid production.
Androgen excess is present in all patients and manifests with broad phenotypic variability, ranging from severe virilization of the external genitalia in neonatal girls (e.g., 46,XX disordered sex development [DSD]) to hirsutism and oligomenorrhea resembling a polycystic ovary syndrome phenotype in young women with nonclassic CAH. In countries without neonatal screening for CAH, boys with classic CAH usually present with life-threatening adrenal crisis in the first few weeks of life (salt-wasting crisis); a simple-virilizing genotype manifests with precocious pseudo-puberty and advanced bone age in early childhood, whereas men with nonclassic CAH are usually detected only through family screening.
Glucocorticoid treatment is more complex than for other causes of primary adrenal insufficiency as it not only needed to replace missing glucocorticoids but also to control the increased ACTH drive and subsequent androgen excess. Current treatment is hampered by the lack of glucocorticoid preparations that mimic the diurnal cortisol secretion profile, resulting in a prolonged period of ACTH stimulation and subsequent androgen production during the early morning hours. In childhood, optimization of growth and pubertal development are important goals of glucocorticoid treatment, in addition to prevention of adrenal crisis and treatment of 46,XX DSD. In adults, the focus shifts to preserving fertility and preventing side effects of glucocorticoid overtreatment, namely, the metabolic syndrome and osteoporosis. Fertility can be compromised in women due to oligomenorrhea/amenorrhea with chronic anovulation as a consequence of androgen excess. Men may develop so-called testicular adrenal rest tumors (Fig. 406-17). These consist of hyperplastic cells with adrenocortical characteristics located in the rete testis and should not be confused with testicular tumors. Testicular adrenal rest tissue can compromise sperm production and induce fibrosis that may be irreversible.
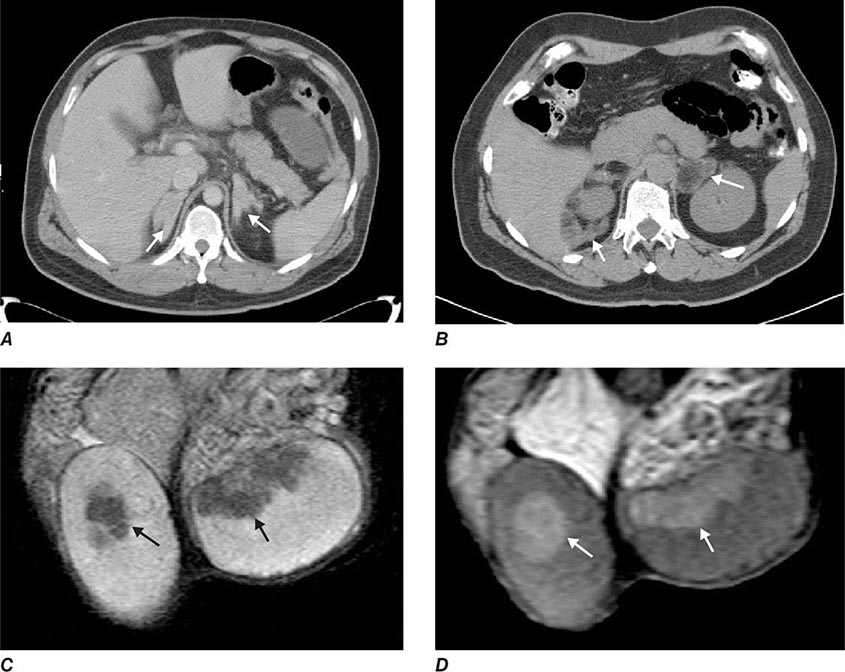
FIGURE 406-17 Imaging in congenital adrenal hyperplasia (CAH). Adrenal computed tomography scans showing homogenous bilateral hyperplasia in a young patient with classic CAH (A) and macronodular bilateral hyperplasia (B) in a middle-aged patient with classic CAH with longstanding poor disease control. Magnetic resonance imaging scan with T1-weighted (C) and T2-weighted (D) images showing bilateral testicular adrenal rest tumors (arrows) in a young patient with salt-wasting congenital adrenal hyperplasia. (Courtesy of N. Reisch.)
TREATMENT | CONGENITAL ADRENAL HYPERPLASIA |
Hydrocortisone is a good treatment option for the prevention of adrenal crisis, but longer acting prednisolone may be needed to control androgen excess. In children, hydrocortisone is given in divided doses at 1–1.5 times the normal cortisol production rate (about 10–13 mg/m2 per day). In adults, if hydrocortisone does not suffice, intermediate-acting glucocorticoids (e.g., prednisone) may be given, using the lowest dose necessary to suppress excess androgen production. For achieving fertility, dexamethasone treatment may be required, but should be only given for the shortest possible time period to limit adverse metabolic side effects. Biochemical monitoring should include androstenedione and testosterone, aiming for the normal sex-specific reference range. 17-Hydroxyprogesterone (17OHP) is a useful marker of overtreatment, indicated by 17OHP levels within the normal range of healthy controls. Glucocorticoid overtreatment may suppress the hypothalamic-pituitary-gonadal axis. Thus, treatment needs to be carefully titrated against clinical features of disease control. Stress dose glucocorticoids should be given at double or triple the daily dose for surgery, acute illness, or severe trauma. Poorly controlled CAH can result in adrenocortical hyperplasia, which gave the disease its name, and may present as macronodular hyperplasia subsequent to long-standing ACTH excess (Fig. 406-17). The nodular areas can develop autonomous adrenal androgen production and may be unresponsive to glucocorticoid treatment.
Mineralocorticoid requirements change during life and are higher in children, explained by relative mineralocorticoid resistance that diminishes with ongoing maturation of the kidney. Children with CAH usually receive mineralocorticoid and salt replacement. However, young adults with CAH should undergo reassessment of their mineralocorticoid reserve. Plasma renin should be regularly monitored and kept within the upper half of the normal reference range.
407 | Pheochromocytoma |
Pheochromocytomas and paragangliomas are catecholamine-producing tumors derived from the sympathetic or parasympathetic nervous system. These tumors may arise sporadically or be inherited as features of multiple endocrine neoplasia type 2, von Hippel–Lindau disease, or several other pheochromocytoma-associated syndromes. The diagnosis of pheochromocytomas identifies a potentially correctable cause of hypertension, and their removal can prevent hypertensive crises that can be lethal. The clinical presentation is variable, ranging from an adrenal incidentaloma to a hypertensive crisis with associated cerebrovascular or cardiac complications.
EPIDEMIOLOGY
Pheochromocytoma is estimated to occur in 2–8 of 1 million persons per year, and ∼0.1% of hypertensive patients harbor a pheochromocytoma. The mean age at diagnosis is ∼40 years, although the tumors can occur from early childhood until late in life. The classic “rule of tens” for pheochromocytomas states that ∼10% are bilateral, 10% are extra-adrenal, and 10% are malignant.
ETIOLOGY AND PATHOGENESIS
Pheochromocytomas and paragangliomas are well-vascularized tumors that arise from cells derived from the sympathetic (e.g., adrenal medulla) or parasympathetic (e.g., carotid body, glomus vagale) paraganglia (Fig. 407-1). The name pheochromocytoma reflects the black-colored staining caused by chromaffin oxidation of catecholamines; although a variety of terms have been used to describe these tumors, most clinicians use this designation to describe symptomatic catecholamine-producing tumors, including those in extra-adrenal retroperitoneal, pelvic, and thoracic sites. The term paraganglioma is used to describe catecholamine-producing tumors in the skull base and neck; these tumors may secrete little or no catecholamine. In contrast to common clinical parlance, the World Health Organization (WHO) restricts the term pheochromocytoma to adrenal tumors and applies the term paraganglioma to tumors at all other sites.
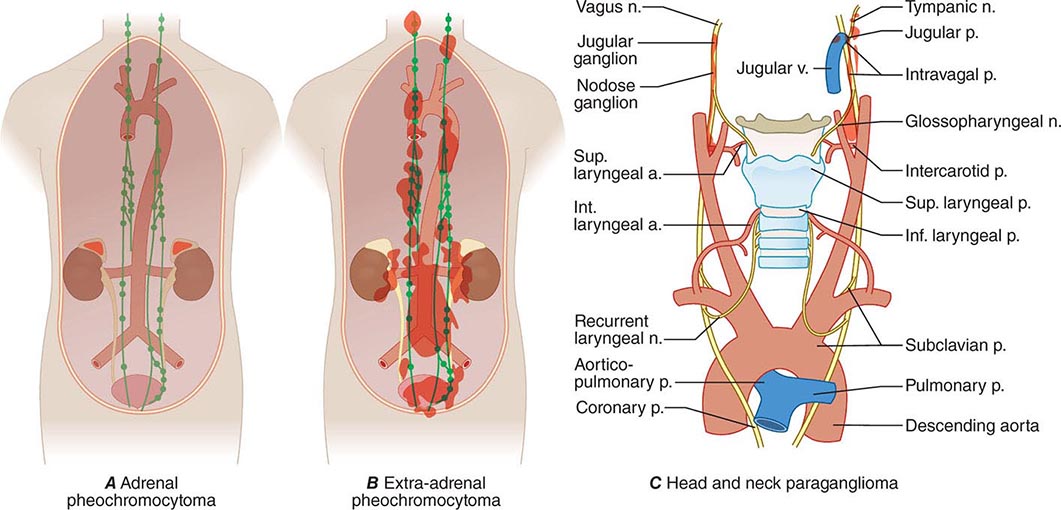
FIGURE 407-1 The paraganglial system and topographic sites (in red) of pheochromocytomas and paragangliomas. (Parts A and B from WM Manger, RW Gifford: Clinical and experimental pheochromocytoma. Cambridge, Blackwell Science, 1996; Part C from GG Glenner, PM Grimley: Tumors of the Extra-adrenal Paraganglion System [Including Chemoreceptors], Atlas of Tumor Pathology, 2nd Series, Fascicle 9. Washington, DC, AFIP, 1974.)
The etiology of sporadic pheochromocytomas and paragangliomas is unknown. However, 25–33% of patients have an inherited condition, including germ-line mutations in the classically recognized RET, VHL, NF1, SDHB, SDHC, and SDHD genes or in the more recently recognized SDHA, SDHAF2, TMEM127, and MAX genes. Biallelic gene inactivation has been demonstrated for the VHL, NF1, and SDH genes, whereas RET mutations activate receptor tyrosine kinase activity. SDH is an enzyme of the Krebs cycle and the mitochondrial respiratory chain. The VHL protein is a component of a ubiquitin E3 ligase. VHL mutations reduce protein degradation, resulting in upregulation of components involved in cell cycle progression, glucose metabolism, and oxygen sensing.
CLINICAL FEATURES
Its clinical presentation is so variable that pheochromocytoma has been termed “the great masquerader” (Table 407-1). Among the presenting manifestations, episodes of palpitation, headache, and profuse sweating are typical, and these manifestations constitute a classic triad. The presence of all three manifestations in association with hypertension makes pheochromocytoma a likely diagnosis. However, a pheochromocytoma can be asymptomatic for years, and some tumors grow to a considerable size before patients note symptoms.
CLINICAL FEATURES ASSOCIATED WITH PHEOCHROMOCYTOMA, LISTED BY FR£EQUENCY OF OCCURRENCE |
The dominant sign is hypertension. Classically, patients have episodic hypertension, but sustained hypertension is also common. Catecholamine crises can lead to heart failure, pulmonary edema, arrhythmias, and intracranial hemorrhage. During episodes of hormone release, which can occur at widely divergent intervals, patients are anxious and pale, and they experience tachycardia and palpitations. These paroxysms generally last <1 h and may be precipitated by surgery, positional changes, exercise, pregnancy, urination (particularly with bladder pheochromocytomas), and various medications (e.g., tricyclic antidepressants, opiates, metoclopramide).
DIAGNOSIS
The diagnosis is based on documentation of catecholamine excess by biochemical testing and localization of the tumor by imaging. These two criteria are of equal importance, although measurement of catecholamines or metanephrines (their methylated metabolites) is traditionally the first step in diagnosis.
Biochemical testing Pheochromocytomas and paragangliomas synthesize and store catecholamines, which include norepinephrine (noradrenaline), epinephrine (adrenaline), and dopamine. Elevated plasma and urinary levels of catecholamines and metanephrines form the cornerstone of diagnosis. The characteristic fluctuations in the hormonal activity of tumors results in considerable variation in serial catecholamine measurements. However, most tumors continuously leak O-methylated metabolites, which are detected by measurement of metanephrines.
Catecholamines and metanephrines can be measured by different methods, including high-performance liquid chromatography, enzyme-linked immunosorbent assay, and liquid chromatography/mass spectrometry. When pheochromocytoma is suspected on clinical grounds (i.e., when values are three times the upper limit of normal), this diagnosis is highly likely regardless of the assay used. However, as summarized in Table 407-2, the sensitivity and specificity of available biochemical tests vary greatly, and these differences are important in assessing patients with borderline elevations of different compounds. Urinary tests for metanephrines (total or fractionated) and catecholamines are widely available and are used commonly for initial evaluation. Among these tests, those for the fractionated metanephrines and catecholamines are the most sensitive. Plasma tests are more convenient and include measurements of catecholamines and metanephrines. Measurements of plasma metanephrine are the most sensitive and are less susceptible to false-positive elevations from stress, including venipuncture. Although the incidence of false-positive test results has been reduced by the introduction of newer assays, physiologic stress responses and medications that increase catecholamine levels still can confound testing. Because the tumors are relatively rare, borderline elevations are likely to represent false-positive results. In this circumstance, it is important to exclude dietary or drug-related factors (withdrawal of levodopa or use of sympathomimetics, diuretics, tricyclic antidepressants, alpha and beta blockers) that might cause false-positive results and then to repeat testing or perform a clonidine suppression test (i.e., the measurement of plasma normetanephrine 3 h after oral administration of 300 μg of clonidine). Other pharmacologic tests, such as the phentolamine test and the glucagon provocation test, are of relatively low sensitivity and are not recommended.
BIOCHEMICAL AND IMAGING METHODS USED FOR DIAGNOSIS OF PHEOCHROMOCYTOMA AND PARAGANGLIOMA |
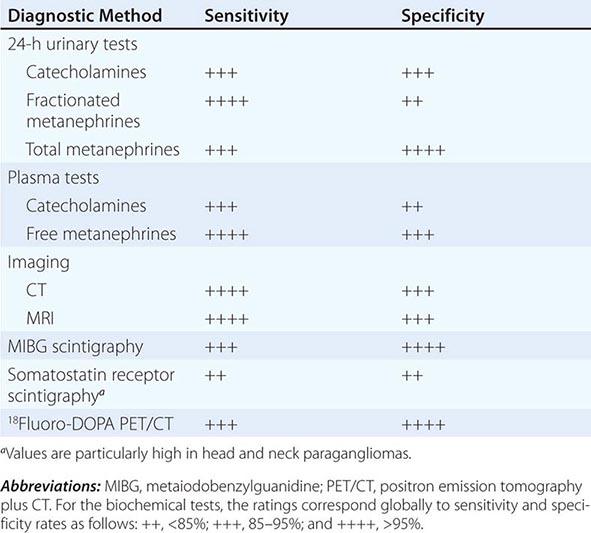
Diagnostic Imaging A variety of methods have been used to localize pheochromocytomas and paragangliomas (Table 407-2). CT and MRI are similar in sensitivity and should be performed with contrast. T2-weighted MRI with gadolinium contrast is optimal for detecting pheochromocytomas and is somewhat better than CT for imaging extraadrenal pheochromocytomas and paragangliomas. About 5% of adrenal incidentalomas, which usually are detected by CT or MRI, prove to be pheochromocytomas upon endocrinologic evaluation.
Tumors also can be localized by procedures using radioactive tracers, including 131I- or 123I-metaiodobenzylguanidine (MIBG) scintigraphy, 111In-somatostatin analogue scintigraphy, 18F-DOPA positron emission tomography (PET), or 18F-fluorodeoxyglucose (FDG) PET. Because these agents exhibit selective uptake in paragangliomas, nuclear imaging is particularly useful in the hereditary syndromes.
Differential Diagnosis When the possibility of a pheochromocytoma is being entertained, other disorders to consider include essential hypertension, anxiety attacks, use of cocaine or amphetamines, mastocytosis or carcinoid syndrome (usually without hypertension), intracranial lesions, clonidine withdrawal, autonomic epilepsy, and factitious crises (usually from use of sympathomimetic amines). When an asymptomatic adrenal mass is identified, likely diagnoses other than pheochromocytoma include a nonfunctioning adrenal adenoma, an aldosteronoma, and a cortisol-producing adenoma (Cushing’s syndrome).
TREATMENT | PHEOCHROMOCYTOMA |
Complete tumor removal, the ultimate therapeutic goal, can be achieved by partial or total adrenalectomy. It is important to preserve the normal adrenal cortex, particularly in hereditary disorders in which bilateral pheochromocytomas are most likely. Preoperative preparation of the patient is important. Before surgery, blood pressure should be consistently below 160/90 mmHg. Classically, blood pressure has been controlled by α-adrenergic blockers (oral phenoxybenzamine, 0.5–4 mg/kg of body weight). Because patients are volume-constricted, liberal salt intake and hydration are necessary to avoid severe orthostasis. Oral prazosin or intravenous phentolamine can be used to manage paroxysms while adequate alpha blockade is awaited. Beta blockers (e.g., 10 mg of propranolol three or four times per day) can then be added. Other antihypertensives, such as calcium channel blockers or angiotensin-converting enzyme inhibitors, have also been used effectively.
Surgery should be performed by teams of surgeons and anesthesiologists with experience in the management of pheochromocytomas. Blood pressure can be labile during surgery, particularly at the outset of intubation or when the tumor is manipulated. Nitroprusside infusion is useful for intraoperative hypertensive crises, and hypotension usually responds to volume infusion.
Minimally invasive techniques (laparoscopy or retroperitoneoscopy) have become the standard approaches in pheochromocytoma surgery. They are associated with fewer complications, a faster recovery, and optimal cosmetic results. Extra-adrenal abdominal and most thoracic pheochromocytomas also can also be removed endoscopically. Postoperatively, catecholamine normalization should be documented. An adrenocorticotropic hormone test should be used to exclude cortisol deficiency when bilateral adrenal cortex–sparing surgery has been performed.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


