8.1 Introduction
Unlike the minimal trial-and-error approach, a systematic drug-product development process (also known as “quality by design,” QbD) includes prior knowledge, the results of experimental designs, quality risk management, and vigilance during the product lifecycle. Such a systematic approach enhances final quality by including understanding and refinement of the formulation and manufacturing process in product development. Thus, the quality of an inhalation product has to be constructed during product and manufacturing-process development in order to meet patient needs and product performance.
The pharmaceutical development phase of a medicinal product’s lifecycle (from idea to market) is when the product and the manufacturing process are designed, with the aim of consistently delivering the intended quality and performance [1, 2]. An inhalation drug product is composed of at least one active pharmaceutical ingredient (API), a number of excipients—for formulation of the API and manufacture of the product—and a primary container (container closure system). The product quality and performance result from the integration and/or interaction of all elements after “assembly.” However, it must be emphasized that the performance of an inhalation product is strongly influenced by the way in which it is manipulated, used, and stored once in the hands of patients and health care professionals.
For every inhalation product, compendial texts such as pharmacopoeias and/or reference guidelines issued by regulatory agencies (European Medicine Agency (EMA), US Food and Drug Administration (FDA)) indicate the characteristics it must comply with to be used effectively and safely. Product characteristics, once identified and stated, enter as specifications in the main section of the product registration dossier (common technical document, CTD).
The characteristics of the product are identified and studied initially during the pharmaceutical development phase, following a rational design. These characteristics refer to drug substances, excipients, formulation, dosage form, container closure system, microbiological attributes, manufacturing process, and instructions for use—all critical to product quality. During pharmaceutical development, every critical formulation attribute and process parameter is studied for the extent to which its variation can impact on the quality of the drug product.
Before looking at the specific studies requested for inhalation products, it should be stated that for all drug products the correct pharmaceutical development strategy focuses on:
- components of the drug product (drug substance and excipients);
- the drug product (formulation development, overages, physicochemical and biological properties);
- manufacturing-process development;
- the container closure system;
- microbiological attributes;
- compatibility with reconstitution diluents (e.g. precipitation, stability) over the recommended in-use shelf life, at the recommended storage temperature.
The drug substance and excipients are studied for those aspects that influence the activity, safety, stability, bioavailability, and manufacturability of a drug product. Physicochemical and biological properties of the drug substance, such as solubility, water content, solid state, particle size, permeability, biological activity, and compatibility with excipients or other drug substances included in the same product, are evaluated. The type, concentration, and properties of excipients are also assessed, relative to their function. Tests are conducted to demonstrate their capacity to function as intended (e.g. as solvents, antioxidants, lubricants, penetration enhancers, release-controlling agents) throughout the drug product’s shelf life.
As for the drug product, formulation development considers those attributes critical for the intended use and route of administration. These studies lead to the evolution of the formulation from the initial concept up to the final design, so long as the properties of drug substance, excipients, container closure system, dosing device, and manufacturing process have been optimized. In this phase, the use of an overage of drug substance (i.e. an amount of drug substance in the finished product higher than the labeled dose) is established and justified on safety and efficacy grounds. The overage can be requested to compensate for expected drug-substance losses during manufacture or product use (e.g. labeled numbers of doses in pressurized metered-dose inhalers (pMDIs) and device-metered dry powder inhalers (DPIs)).
Drug-product development studies allow for identification of the physicochemical and biological properties relevant to product safety and performance, including formulation attributes and manufacturability. For example, studies have to include the development of a test for the respirable fraction (fine-particle mass) of an inhalation product.
The rationale for selecting the container closure system is based on the type of drug product and the suitability of the device for storage and transportation (compatibility with the content). In this regard, specific studies are conducted to justify the choice of materials for primary packaging, demonstrating the integrity of the container and closure. If the container closure includes a dosing device (e.g. DPI), studies are required to demonstrate that an accurate and reproducible dose of the product is delivered under testing conditions simulating the use of the product by patients.
Finally, microbiological attributes of the drug product must be considered during pharmaceutical development with respect to microbial limits for nonsterile products, the effectiveness and safety of preservative systems, sterility maintenance, and microbial challenge testing under conditions simulating patient use.
Development of the manufacturing process starts with consideration of the critical formulation attributes and the available manufacturing-process options, in order to select suitable process components and equipment. This allows any critical process parameter that should be monitored or controlled to be identified, in order to ensure that the product will have the desired quality. In general, development studies are conducted on more than one batch, thus accounting for batch variability.
In conclusion, the results obtained from pharmaceutical development studies combined with manufacturing controls create the so-called “product design space”, that is the multidimensional combination/interaction of input variables (material attributes and process parameters) and output variables (product quality responses). Product variations within the design space are expected to remain under control.
The amount of data collected will be used to support the specifications proposed for the product, to be assessed routinely with every batch manufactured. Moreover, the development studies guarantee that those performance characteristics of the product that will not be routinely tested have been adequately investigated.
8.2 Pharmaceutical Development Studies for Inhalation Products
Inhalation drug products are “human medicinal products intended for delivery of the drug substance into the lungs, or to the nasal mucosa, with the purpose of evoking a local or systemic effect” [3, 4]. These relatively complex means of dosage are typical combination products, comprising the formulation and a delivery device (Figure 8.1). The two combined elements “work together” for the efficacy of the therapy.

Inhalation therapy is successful so long as the active substance enters the airways at the expected dose and deposits on the respiratory mucosa. Upon deposition, a solid drug substance dissolves, is locally absorbed, and either exerts its effect onsite or becomes available for systemic distribution [5, 6]. As the user activates the device, the drug formulation is transformed into a respirable aerosol of liquid droplets or solid particles, which can enter the airways by inhalation. The size distribution of the aerosol particles/droplets is the key characteristic determining the deposition site in the airways.
The delivery device coupled to the specific formulation is responsible for proper aerosol generation. In addition, in the case of multidose products, the delivery device also meters the amount of drug formulation that leaves the device at actuation. Hence, dose-metering and aerosol generation are two steps peculiar to inhalation-drug-product performance and are the focus of related development studies. Consequently, regulatory agencies demand with respect to the dose emitted (i.e. the amount of drug exiting the device and available to the patient) and the size distribution of the aerosol mist (i.e. the aerosol respirability and the deposition site in the airways).
Inhalation drug products are categorized in four main types: pressurized metered-dose inhalers (pMDIs), DPIs, products for nebulization (single-dose or multidose), and metered-dose nebulizers (MDNs). DPIs can be further classified, as the dose can either be metered by the device at actuation or premetered during manufacture.
pMDIs consist of a drug substance formulated in suspension or solution in a liquefied propellant, stored in a metal canister sealed by a crimped metering valve, such that internally the formulation is under pressure (Figure 8.2). This internal pressure gives the energy needed to deliver and aerosolize the drug dose as the metering chamber is opened to the atmosphere; the propellant rapidly evaporates, producing the fine mist of solid particles or liquid droplets (depending on the formulation) that the patient inhales [7].
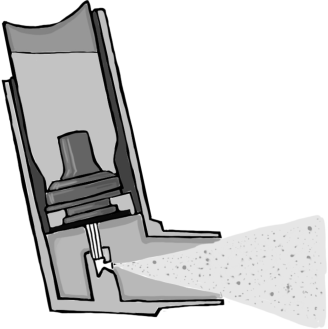
DPIs deliver a solid powder to the lungs [8, 9]. The powder is either a micronized drug substance or, more commonly, a blend of micronized drug and larger carrier particles (such as lactose). In either case, the powder is delivered by means of a device in which the energy for delivery and aerosolization comes from the inspiratory flow of the patient’s inhaling through the device.
Liquids for inhalation (solutions or suspensions) required “coupling” to an aerosolizing device (nebulizer) at the time of use. The nebulizer design and nebulization mechanism actually affect droplet size distribution, delivery rate, and total dose delivered. Since the formulation and device are often developed independently and various nebulizers are available on the market, the actual dose delivered to the patient remains uncertain and highly variable. However, in recent years technological improvements have been seen in this field, and the use of aerosolization in aqueous carrier is being reconsidered [10].
Given the variety of inhalation products and the complexity of their proper use and effectiveness, their pharmaceutical development requires many factors to be investigated and understood. Table 8.1, reproduced from EMA guidelines, provides a list of tests required to characterize inhalation products during pharmaceutical development [3].
Table 8.1 Pharmaceutical development studies for inhalation products. White lines: studies demonstrating the product performance with respect to formulation, delivery device, and their combination; dark grey lines: studies demonstrating the product performance in the hands of the patient (handling, use, storage); light grey lines: additional studies for specific development requirements.
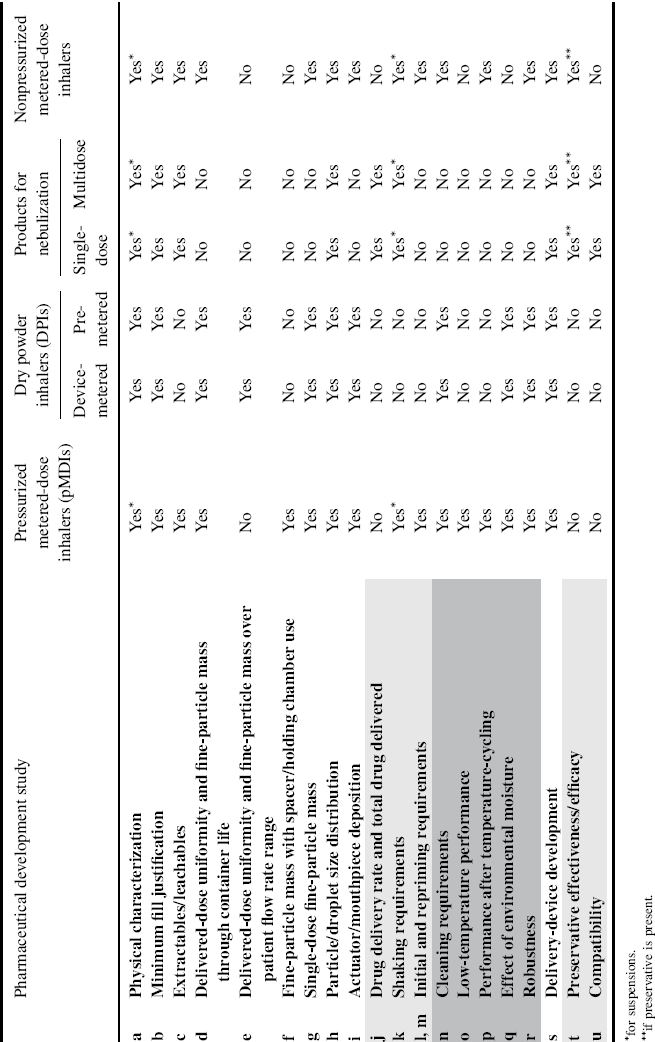
As shown in the table, it is not necessary to perform all tests on every product. Some of them are applicable only to particular products (e.g. low-temperature performance for pMDIs) and some depend on specific label instructions for use (e.g. shaking requirements for products in suspension). It is possible to subdivide these pharmaceutical development studies into three main groups:
8.2.1 Studies Demonstrating Product Performance with Respect to Formulation, Delivery Device, and their Combination
Physical Characterization
The physical properties of the drug substance and excipients may be relevant to the safety and efficacy of the product. These properties are solubility, particle size and morphology, density, surface rugosity, charge, and crystallinity [11–13]. For example, the dissolution of the drug substance in pMDI liquefied propellants makes a difference in product performance with respect to the particle size of the generated aerosol [14, 15]. In fact, if the formulation is a suspension, the aerosol mist emitted upon valve actuation is composed of propellant droplets containing solid drug particles, whose size is expected to be the same established at manufacture. As the propellant evaporates, the solid drug particles already have a size suitable for inhalation and airway deposition. In contrast, if a drug solution is emitted, solid drug particles will be generated as a result of propellant evaporation, and their size (and shape) will depend on propellant evaporation rate, the presence of cosolvents, drug solubility, and so on. Clearly, this last process must be controlled to ensure that the size characteristics and degree of crystallinity of the newly formed drug particles meet the requirements for inhalation (respirability).
Particle size, shape, and density are combined in the definition of the aerodynamic diameter (dae). This equivalent spherical diameter describes the capability of aerosol particles to fly in the inhaled airstream, enter the airways, and deposit at a specific site [16, 17]. Hence, size, shape, and density define the aerodynamic behavior of solid particles—either drug or excipient. In addition, for inhalation powders, particle size, shape, and density affect the flow and packing properties of the particle collection—that is, the powder. Dosage-form manufacture (drug/carrier blending, device-filling for multidose DPIs, capsule- or blister-filling for premetered single-dose devices) and product use (dose-metering, ejection from metering chamber/capsule, aerosolization) are strongly influenced by these fundamental and derived properties of the powder.
Drug particles for inhalation must be micronized with an aerodynamic diameter in the range 1–5 μm for respirability and lung deposition. However, the small size makes them poorly flowable, highly cohesive, and prone to aggregation. These drawbacks complicate the manufacturing phase while capsules or blisters are being filled with the correct drug dose, which by the way is often very low for inhaled APIs (microgram to milligram range). Moreover, a cohesive powder may be only partially aerosolized and emitted from the inhaler, thus causing a fraction of the dose to be unavailable to the patient. Again, if microparticles form aggregates of variable size, powder respirability and deposition will change, with unpredictable effects on drug bioavailability.
These are the reasons why flow properties should be improved, using several approaches [18–20] which change the physical appearance of the inhalation powders. For instance, many DPI formulations contain mixtures of micronized drug particles and larger lactose carrier particles, as shown in Figure 8.3. The carrier improves powder-handling, dosing, and aerosolization [21]. Then, immediately upon inhalation, the blend is deaggregated due to the turbulence of the inhaled airstream: the micronized drug particles enter the airways, while carrier particles deposit in the mouth.
Figure 8.3 Scanning electron microscope microphotograph of an ordered mixture of lactose carrier particles and micronized drug particles. Courtesy Dr Francesca Buttini, Department of Pharmacy, University of Parma, Parma, Italy

In alternative to ordered mixtures, highly respirable “engineered particles” can be designed by playing with their geometric size, shape, and density in order to define their aerodynamic size.
Whatever technology is chosen, the physical properties of drug particles, such as size, shape, density, and rugosity, will have to be assessed by means of suitable analytical tests (e.g. laser light scattering, electron microscopy, atomic force microscopy, etc.) during pharmaceutical development.
Finally, when a solid aerosol is inhaled, dissolution at the deposition site must occur for the active substance to become bioavailable. Thus, physical characterization must also consider solubility and crystallinity, as these may affect the rate of the dissolution process.
Minimum Fill Justification
Inhalation products exist in single-dose or multidose versions. Among multidose versions, the dose can be either premetered into a reservoir during manufacture or metered by the device at the time of use. Typical examples of multidose products are pMDIs and device-metered DPIs. These precisely meter a defined number of unit doses at each actuation. The number of available doses per container must be stated on the product label. During product use, the device indicates to the patient how many doses are still available by means of a dose counter [22]. The total amount of formulation (liquid or solid) to be filled into the container must be sufficient to provide the labeled number of actuations and is defined during drug-product development. This individual-container minimum fill guarantees that every dose of the total number labeled (particularly the final ones, delivered when the container is almost empty) meets the drug-product specification limits for delivered-dose uniformity and fine-particle mass (see the following sections).
Consequently, during product development, a study will be conducted to demonstrate the suitability of the individual-container minimum fill adopted. The study should prove that the filled amount is the minimum required to deliver all labeled doses. The extra fill added for the uniformity of the final doses must not induce the patient into prolonging use after delivering the last counted dose.
For premetered DPIs and products for nebulization, each unit dose is premetered by the manufacturer in a proper reservoir (capsule, blister, vial, ampoule), and the number of unitary containers in the box determines the labeled number of doses. In this case, the fill volume and/or weight inside the capsule or vial should be justified to comply with the specifications of delivered-dose uniformity and fine-particle mass.
Extractables/Leachables
Determining the profiles of extractable and leachable compounds means evaluating any possible physicochemical interactions between the formulation and the container closure system [23]. The direct contact between content and container might lead to the presence in the drug product of “foreign” substances from the container. In the worst case, the drug product might be adulterated by a substance contained in the container (or a part of it) which renders it harmful. In this regard, regulatory expectations are higher for inhalation products than for other medicinal categories (e.g. topical, oral), as higher toxicological concerns relate to substance delivery to the lungs.
As a general definition, “extractables” are all compounds present in the container closure system that can be potentially extracted from it, independently of the drug product filled in. This means that extractables are specific to the materials (plastic, rubber) of which the container closure is made. On the other hand, “leachables” are a subset of extractables which can pass into the formulation under normal storage conditions during shelf life. Given a certain container closure, leachables will then be specific to each drug formulation.
Among the various types of inhalation product, the highest probability of an interaction exists in pMDIs, due to the presence of propellants and cosolvents that can extract into the formulation organic compounds from the valve or the canister coating. Therefore, assessing and quantifying the presence of leachables at trace levels (i.e. parts per million or billion) is a fundamental requirement during pMDI development. The issue is less relevant to DPIs, as an interaction between solids is rather unlikely to occur.
Packaging materials are selected at an early stage of product development, especially in the case of inhalation products where the container closure system contains and delivers the formulation. As for pharmaceutical excipients, for containers and closures compendial or noncompendial plastic and rubber materials are available to the drug-product manufacturer. Compendial materials are described in official pharmacopoeias. For this reason, their manufacturers (or the container manufacturers) are responsible for guarantying and certifying compliance with the specifications relevant for the intended use (including limits for extractables). Nevertheless, the justification and proof of any container-closure applicability is the responsibility of the drug-product applicant. It is required that the leachables profile for the compendial plastic and rubber components of the container closure selected for a certain formulation is determined.
For noncompendial materials that are in contact with the formulation (e.g. valves), the extractables profile will be determined by the drug-product applicant, and the conditions/results of the study will be provided in detail (e.g. solvents used, temperature, storage time). In addition, it is important to determine whether any of the extractables are also leachables found in the formulation at the end of the product shelf life or when the equilibrium point is reached.
A compound that appears as leachable should be identified and have its safety assessed based on suitable safety thresholds. Unfortunately, typical leachables “contaminate” the formulation at trace levels and the analytical procedures for their accurate identification and quantification can be complicated. In contrast, extractables can be present in the order of parts per thousand to per cent on the basis of material weight, and can thus be more easily detected, with positive results.
Delivered-Dose Uniformity and Fine-Particle Mass Through Container Life
The delivered dose is the amount of drug emitted in the form of an aerosol mist from the inhaler upon device actuation and available for inhalation.
The respirability of the aerosolized preparation is evaluated by the aerodynamic assessment of fine particles using a cascade impactor (see Chapter 6). The aerosol particles to be inhaled in the delivered dose must be formulated in such a way that a significant amount enters the lung. This part of the delivered dose is called the fine-particle mass and represents the amount of emitted aerosol particles suitable for lung deposition (i.e. with an aerodynamic size <5 μm) [24].
Since the aerosol generated by an inhaler is a collection of particles, it is unlikely they will all have the same size; rather, they will show a more or less narrow size distribution, in the micrometer range. Basically, the fine-particle mass measures the respirable dose of the inhalation product. Development studies are aimed at improving the respirability of the inhalation product by increasing the fraction with a size suitable for deposition following inhalation. During pharmaceutical development, both delivered dose and fine-particle mass are studied in detail in order to guarantee their consistency according to drug-product application [25]. The goal is to minimize the difference between delivered dose and fine-particle mass.
The delivered-dose uniformity is experimentally measured by actuating the device a defined number of times and quantitatively collecting the emitted product into a specific sampling apparatus (see Chapter 6). The amount of drug in the sample is then quantified by a suitable analytical method (e.g. high-performance liquid chromatography, HPLC). The test is always conducted on the minimum delivered dose, which in some cases is emitted with more than one actuation.
Delivered dose and fine-particle mass are studied throughout the container life, from the first dose until the last labeled. The product performance must remain consistent regardless of whether the container is 100% full, half empty, or close to exhaustion. In general, a total of at least 10 doses has to be analyzed, combined from the beginning, middle, and end of the container’s life.
For products which require priming before first use, the priming procedure must be studied and established to guarantee consistency of the first emitted dose. The product containers have to be handled and tested in the same way in which a patient would use them. Storage orientation, cleaning instructions, and dosing interval have to be considered. Moreover, a study should be conducted on the doses between the last labeled and the very last one at exhaustion. The rationale is to characterize the tail-off profile in case of incidental product use beyond the labeled number of doses (this test is not required if the product has a lock-out mechanism preventing such misuse).
Studies of delivered-dose uniformity and fine-particle mass are performed for all inhalation products designed to deliver a metered drug dose at each actuation (i.e. not for products for nebulization).
Delivered-Dose Uniformity and Fine-Particle Mass Over Patient Flow Rate Range
Delivered-dose uniformity and fine-particle mass must be studied in order to identify all variables that may affect them during product use. One main variable is the respiratory capacity of the patient, expressed in terms of inspiratory flow rate.
In the case of products for nebulization and pMDIs, the inspiratory airflow only captures the emitted aerosol, as the nebulizer and the propellant provide dose aerosolization and delivery. Hence, patient respiratory capacity is not an issue for delivered-dose uniformity and fine-particle mass, which only depend on the functioning mechanism of the device. Consequently, the product development is performed under a fixed airstream rate through the measuring apparatus.
In contrast, DPIs are devices in which the energy for dose aerosolization and delivery is given by the inspiratory act of the patient inhaling through the device. As the respiratory capacity and inhalation rate vary from patient to patient, this source of variability in product performance has to be addressed [26]. Hence, for DPIs, uniformity of delivered dose and fine-particle mass have to be studied over the range of flow rates achievable through the delivery device by the intended patient population. The device itself is also a variable to consider in defining the experimental conditions of this study, as its design determines the resistance offered to the act of inhalation through it. Consequently, for each device, a range of flow rates between 28 and 100 L/min must be identified (minimum, median, and maximum achievable) and justified based on clinical or published data. If the minimum flow rate is not sufficient to produce an acceptable delivered dose, information should be provided to health care professionals about the effect of inspiratory flow rate on product performance.
Fine-Particle Mass with Spacer/Holding Chamber Use
The spacer is a chamber mounted at the exit of the actuator of a delivery device that allows for the reduction of the aerosol emission speed in order to facilitate product use by the patient (Figure 8.4). The effect on fine-particle mass of the presence of a spacer/holding chamber only needs to be investigated for pMDIs requiring such an accessory. The spacer helps patients, particularly children and elderly people, in synchronizing device-firing with the inhalation act [27, 28].
Figure 8.4 Spacers for children (left) and adults (right). Courtesy of Chiesi Farmaceutici, Parma, Italy

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


