Pharmaceutical applications of microbiological techniques
Norman A. Hodges
Chapter contents
Measurement of antimicrobial activity
Factors to be controlled in the measurement of antimicrobial activity
Minimum inhibitory concentration determinations (MICs)
Preservative efficacy tests (PETs or challenge tests)
Microbiological quality of pharmaceutical materials
Key points
Introduction
The purpose of this chapter is to bring together those microbiological methods and procedures that are relevant to the design and production of medicines and medical devices. These are methods used (a) to determine the potency or activity of antimicrobial chemicals, e.g. antibiotics, preservatives and disinfectants, and (b) as part of the microbiological quality control of manufactured sterile and non-sterile products. The chapter describes the experimental procedures that are unique or particularly relevant to pharmacy, rather than those that are common to microbiology as a whole. In the latter category, for example, are procedures used to identify and enumerate microorganisms. These, together with staining and microscopical techniques, are described in Chapter 13.
Several of the methods and tests discussed here are the subject of monographs or appendices in pharmacopoeias or they are described in national and international standards or other recognized reference works. It is not the intention to reproduce these official testing procedures in detail but rather to explain the principles of the tests, to draw attention to difficult or important aspects, and to indicate the advantages, problems or shortcomings of the various methods.
Measurement of antimicrobial activity
In most of the methods used to assess the activity of antimicrobial chemicals, an inoculum of the test organism is added to a solution of the chemical under test, samples are removed over a period of time, the chemical is inactivated and the proportion of surviving cells determined. Alternatively, culture medium is present together with the chemical and the degree of inhibition of growth of the test organism is measured. In each case it is necessary to standardize and control such factors as the concentration of the test organism, its origin, i.e. the species and strain employed, together with the culture medium in which it was grown, the phase of growth from which the cells were taken, and the temperature and time of incubation of the cells after exposure to the chemical. Because such considerations are common to several of the procedures described here, e.g. antibiotic assays, preservative efficacy (challenge) tests and determinations of minimum inhibitory concentration (MIC), it is appropriate that they should be considered first, both to emphasize their importance and to avoid repetition.
Factors to be controlled in the measurement of antimicrobial activity
Origin of the test organism
Although two cultures may bear the same generic and specific name, i.e. they may both be called Escherichia coli, this does not mean that they are identical. Certainly, they would normally be similar in many respects, e.g. morphology (appearance), cultural requirements and biochemical characteristics, but they may exhibit slight variations in some of these properties; such variants are described as strains of E. coli. A variety of strains of a single species may normally be obtained from a culture collection, e.g. the National Collection of Industrial and Marine Bacteria or the National Collection of Type Cultures. Different strains may also occur in hospital pathology laboratories by isolation from swabs taken from infected patients or by isolation from contaminated food, cosmetic or pharmaceutical products, and from many other sources. Strains obtained in these ways are likely to exhibit variations in resistance to antimicrobial chemicals. Strains from human or animal infections are frequently more resistant to antimicrobial chemicals, particularly antibiotics, than those from other sources. Similarly, strains derived from contaminated medicines may be more resistant to preservative chemicals than those obtained from culture collections. Therefore, in order to achieve results that are reproducible by a variety of laboratories, it is necessary to specify the strain of the organism used for the determination.
It is becoming increasingly common, too, for official testing methods to limit the number of times the culture collection specimen may be re-grown in fresh medium (called the number of subcultures or passages) before it must be replaced. This is because the characteristics of the organism (including its resistance to antimicrobial chemicals) may progressively change as a result of mutation and natural selection through the many generations that might arise during months or years of laboratory cultivation.
Composition and pH of the culture medium
There are several methods of assessing antimicrobial activity which all have in common the measurement of inhibition of growth of a test organism when the antimicrobial chemical is added to the culture medium. In such cases the composition and pH of the medium may influence the result. The medium may contain substances that antagonize the action of the test compound, e.g. high concentrations of thymidine or para-aminobenzoic acid will interfere with sulfonamide activity.
The antimicrobial activities of several groups of chemical are influenced by the ease with which they cross the cell membrane and interfere with the metabolism of the cell. This, in turn, is influenced by the lipid solubility of the substance, because the membrane contains a high proportion of lipid and tends to permit the passage of lipid-soluble substances. Many antimicrobial chemicals are weak acids or weak bases, which are more lipid soluble in the unionized form. The pH of the environment therefore affects their degree of ionization, hence their lipid solubility and so, ultimately, their antimicrobial effect. Benzoic acid, for example, is a preservative used in several oral mixtures which has a much greater activity in liquids buffered to an acid pH value than those which are neutral or alkaline. Conversely, the aminoglycoside antibiotics, e.g. streptomycin, neomycin and gentamicin, which are weak bases, are more active at slightly alkaline pH values, although this is more a consequence of the transport systems by which the molecules enter the bacterial cell working better at alkaline pH than of enhanced lipid solubility. The presence of organic matter, e.g. blood, pus or serum, is likely to have a marked protective effect on the test organism and so antimicrobial chemicals may appear less active in the presence of such material. The activity of several antibiotics, notably tetracyclines and aminoglycosides, is reduced by the presence of high concentrations of di- or trivalent cations in the medium.
Exposure and incubation conditions
The temperature, duration and redox conditions of exposure to the antimicrobial chemical (or incubation of survivors after exposure) may all have a significant effect on its measured activity. Increasing the temperature of exposure of the test organism to the chemical increases the antimicrobial activity by a factor which is quantified by the temperature coefficient (Q10 value: the number of times increase in activity for a 10 °C rise in temperature). Phenols and alcohols, for example, may respectively exhibit Q10 values of 3–5 and > 10, and so a variation of 5 °C in the temperature of exposure (which is permitted by pharmacopoeial preservative efficacy tests) may lead to a markedly different rate of kill of the organism in question.
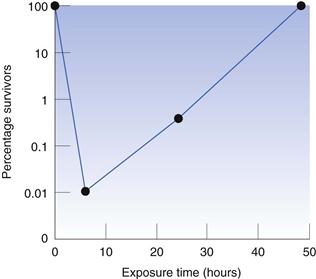
The period of time for which the test organism is exposed to the antimicrobial chemical may influence the recorded result because it is possible for the organism to adapt and become resistant to the presence of the chemical. In preservative efficacy tests, the exposure period is normally 28 days, which is sufficient time for any cells that are not killed during the first 24–48 hours to recover and start to reproduce, so that the final bacterial concentration may be much higher than that at the start. This is illustrated in Figure 14.1, which shows the effect of the quaternary ammonium preservative benzethonium chloride on Pseudomonas aeruginosa. The concentration of bacteria was reduced to approximately 0.01% of the initial value during the first 6 hours, but the bacteria that survived this early period recovered to the original level within 2 days. There is the potential for a similar phenomenon to arise in other situations, e.g. in minimum inhibitory concentration (MIC) determinations of bacteriostatic agents (those that do not kill but merely inhibit the growth of the test organism), although it is not common in MICs because the exposure (incubation) time is much shorter than that in preservative testing.
The effect of some antibiotics may be influenced by the redox conditions during their period of contact with the test organism. Aminoglycosides, for example, are far less active, and metronidazole is far more active, under conditions of low oxygen availability. Such effects may even be seen during agar diffusion antibiotic assays, in which the antibiotic diffuses from a well into an agar gel inoculated with the test organism; the diameter of the zone of growth inhibition that surrounds a well filled with neomycin solution, for example, may be significantly greater at the surface of the agar (where there is abundant oxygen) than at its base, where the oxygen concentration is limited by its poor diffusion through the gel.
Inoculum concentration and physiological state
It is perhaps not surprising that the concentration of the inoculum can markedly affect antimicrobial action, with high inoculum levels tending to result in reduced activity. There are two main reasons for this. First, there is the phenomenon of drug adsorption on to the cell surface or absorption into the interior of the cell. If the number of drug molecules in the test tube is fixed yet the number of cells present is increased, this obviously results in fewer molecules available per cell and consequently the possibility of a diminished effect. In addition to this there is the second, more specialized case, again concerning antibiotics, where it is frequently observed that certain species of bacteria can synthesize antibiotic-inactivating enzymes, the most common of which are the various types of β-lactamases (those destroying penicillin, cephalosporin and related antibiotics). Thus a high inoculum means a high carryover of enzyme with the inoculum cells, or at least a greater potential synthetic capacity.
Perhaps less predictable than the inoculum concentration effect is the possibility of the inoculum history influencing the result. There is a substantial amount of evidence to show that the manner in which the inoculum of the test organism has been grown and prepared can significantly influence its susceptibility to toxic chemicals. Features such as the nature of the culture medium, e.g. nutrient broth or a defined glucose-salts medium, the metal ion composition of the medium and hence of the cells themselves, and the physiological state of the cells, i.e. ‘young’ actively growing cells from the logarithmic growth phase or ‘old’ non-dividing cells from the stationary phase, all have the potential to influence the observed experimental values. Generally, antimicrobial chemicals are more effective against actively growing cells than slowly growing or dormant ones, e.g. bacterial spores.
Antibiotic assays
Methods of assaying antibiotics may be broadly divided into three groups:
Biological methods offer the advantage that the parameter being measured in the assay (growth inhibition) is the property for which the drug is used, and so inactive impurities or degradation products will not interfere and lead to an inaccurate result. Biological methods also offer other advantages (Table 14.1) but they have several significant limitations and non-biological methods are now generally preferred.
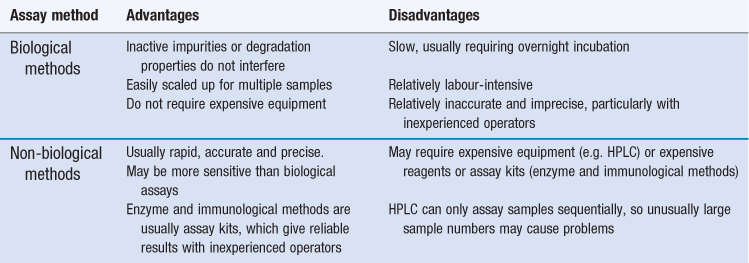
Enzyme-based and immunoassay kits are used in hospitals, notably for therapeutic monitoring of toxic antibiotics (e.g. aminoglycosides and vancomycin), whereas HPLC tends to be preferred in the pharmaceutical industry, particularly for quality assurance applications. Biological assays are most likely to be used when the alternatives are inappropriate, especially when the active antibiotic cannot readily be separated from inactive impurities, degradation products or interfering substances, or it cannot easily be assayed by HPLC without derivatization to enhance ultraviolet absorption (e.g. aminoglycosides). These situations may arise:
Biological antibiotic assays, or bioassays as they are frequently known, may be of two main types: agar diffusion and turbidimetric. The European Pharmacopoeia (PhEur) (2011) describes experimental details for both methods, e.g. test microorganisms, solvents, buffers, culture media and incubation conditions. In each case, a reference material of known activity must be available. When antibiotics were in their infancy, few could be produced in the pure state free from contaminating material, and specific chemical assays were rarely available. Thus the potency or activity of reference standards was expressed in terms of (international) units of activity. There are few antibiotics for which dosage is still normally expressed in units: nystatin and polymyxin are two of the remaining examples. More commonly, potencies are recorded in terms of µg mL−1 of solution or µg antibiotic mg−1 of salt, with dosages expressed in mg. Antibiotic assay results are usually in the form of a potency ratio of the activity of the unknown or test solution divided by that of the standard.
Agar diffusion assays
In this technique, the agar medium in a Petri dish or a larger assay plate is inoculated with the test organism, wells are created by removing circular plugs of agar, and these wells are filled with a solution of the chemical under test (Fig. 14.2).
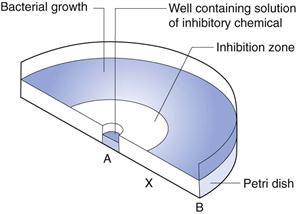
The chemical diffuses through the gel from A towards B and the concentration falls steadily in that direction. The concentration in the region A to X is sufficiently high to prevent growth, i.e. it is an inhibitory concentration. Between X and B the concentration is sub-inhibitory and growth occurs. The concentration at X at the time the zone edge is formed is known as the critical inhibitory concentration (CIC). After incubation, the gel between A and X is clear and that between X and B is opaque as a result of microbial growth which, with the common test organisms, is usually profuse. A zone of inhibition is therefore created, the diameter of which will increase as the concentration of chemical in the well increases.
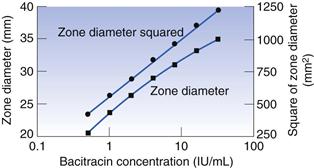
A graph may be constructed which relates zone diameter to the logarithm of the concentration of the solution in the well (Fig. 14.3). It is normally found to be linear over a small concentration range but the square of the diameter must be plotted to achieve linearity over a wide range. A plot such as that in Figure 14.3 may, quite correctly, be used to calculate the concentration of a test solution of antibiotic. In practice, however, it is found to be more convenient to obtain reliable mean zone diameters for the standard at just two or three concentrations, rather than somewhat less reliable values for six or seven concentrations. There is no reason why an assay should not be based upon a two- or three-point line, provided that those points are reliable and that preliminary experiments have shown that the plotted relationship over the concentration range in question is linear.
It is not common to conduct antibiotic assays in Petri dishes because too few zones may be accommodated on a standard-sized dish to permit the replication necessary to obtain the required accuracy and precision. Antibiotic assays, when performed on a large scale, are more often conducted using large assay plates 300 mm or more square (Fig. 14.4). The wells are created in a square design and the number that may be accommodated will depend upon the anticipated zone diameters: 36 or 64 wells are common (6 × 6 or 8 × 8, respectively). The antibiotic standard material may be used in solution at three known concentrations (frequently referred to as ‘doses’) and the antibiotic solution of unknown concentration treated likewise; alternatively, each may be employed at two concentrations. A randomization pattern known as a Latin square is used to ensure that there is a suitable distribution of the solutions over the plate, thereby minimizing any errors due to uneven agar thickness.

Fig. 14.4 Antibiotic agar diffusion assay conducted using a 6 x 6 assay design in a 300 mm square assay plate.
In the case of an assay based upon standard solutions used at two concentrations, the potency ratio may be calculated directly from the graph (as shown in Fig. 14.5) or by using the formula below:
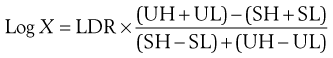 (14.1)
(14.1)
where X is the potency ratio, LDR is the logarithm of the dose ratio (i.e. ratio of concentrations of standard solutions), UH, UL, SH and SL are the mean zone diameters for the unknown and standard high and low doses. The derivation of this is described in detail by Wardlaw (1999), who deals extensively with the subject of antibiotic assays. The tests for acceptable limits of parallelism between the line joining the standards and that joining the test points, together with confidence limits applicable to the calculated potency ratios, are described in the current PhEur.
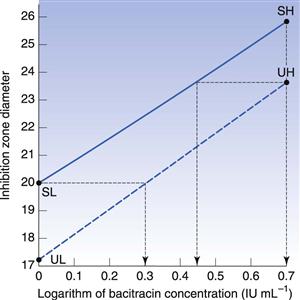
In calculating the potency ratio directly from Figure 14.5, the zone diameters for the standard and unknown high concentrations are plotted at the same abscissa values, and those for the low concentrations similarly. Two zone diameters are considered which are as widely separated on the ordinate as possible while still being covered by the standard and the test lines. The ratio of the concentrations required to achieve the selected diameter is thus an estimate of the potency ratio. The mean of the two estimates taken at the extremes of the range of common zone diameters should be identical to the value by calculation from the formula. Thus, in Figure 14.5, at a zone diameter of 23.75 mm the first estimate of potency ratio is 0.557 (antilog of 0.445 divided by antilog of 0.699); the second is 0.507 (antilog of zero divided by the antilog of 0.295). The mean value of 0.53 indicates the unknown solution to have approximately half the activity of the standard.
Practical aspects of the conduct of agar diffusion assays.
The agar may be surface inoculated or inoculated throughout while in the molten state prior to pouring. In the latter case zones may arise which are different in diameter at the agar surface than at the base of the Petri dish; this may complicate the recording of zone diameters. Zones which are not perfectly circular may be disregarded, although it may be appropriate to record the mean of the long and short axes. Such zones may result from non-circular wells, careless filling or uneven drying of the agar gel owing to a poorly fitting plate cover. The zones may be read directly with callipers or, more conveniently, after enlargement by projection onto a screen. Automatic zone readers incorporating a series of photocells that detect opacity changes at the zone edge are available, and may be linked to a personal computer which rapidly calculates the result together with the appropriate statistical analyses. The size of the zone is determined by the relative rates of diffusion of the drug molecule and growth of the test organism. If the assay plates are left at room temperature for 1–4 hours prior to incubation, growth is retarded whereas diffusion proceeds. This may result in larger zones and improved precision.
The zone diameter is affected by most of the factors previously stated to influence antimicrobial activity and, in addition, gel strength and the presence of other solutes in the antibiotic solution, e.g. buffer salts. If the antibiotic has been extracted from a formulated medicine, e.g. cream, lotion or mixture, excipients may be simultaneously removed and influence the diffusion of the antibiotic in the gel; sugars are known to have this effect. Because antibiotic assays involve a comparison of two solutions that are similarly affected by changes in experimental conditions, day-to-day variations in, for example, inoculum concentration will not have a great effect on the accuracy of the potency ratio obtained. However, the precision may be affected. The volume of liquid in the well is of minimal importance; it is usually of the order of 0.1 mL and is delivered by semi-automatic pipette. As an alternative to wells, the antibiotic may be introduced on to the agar using absorbent paper discs, metal cylinders or ‘fish spine’ beads (beads having a hole drilled in them which contains the liquid).
For many antibiotics, the test organism is a Bacillus species and the inoculum is in the form of a spore suspension, which is easy to prepare, standardize and store. Alternatively, frozen inocula from liquid nitrogen may be used as a means of improving reproducibility.
Careful storage and preparation of the reference standards are essential. The reference antibiotic is usually stored at low temperature in a freeze-dried condition.
Turbidimetric assays
In this case, antibiotic standards at several concentrations are incorporated into liquid media and the extent of growth inhibition of the test organism is measured turbidimetrically using a nephelometer or spectrophotometer. The unknown or test antibiotic preparation is run simultaneously, again at several concentrations, and the degree of growth inhibition compared. Such assays are less commonly used than agar diffusion methods because their precision is rather inferior but they do have the advantage of speed: the result may be available after an incubation period as short as 3–4 hours. They are also more sensitive than diffusion assays and consequently may be applied to low-activity preparations.
The shape and slope of the dose–response plot for a turbidimetric assay may be more variable than that for agar diffusion, and non-linear plots are common. Typical dose–response plots are shown in Hewitt & Vincent (1989). The plotted points are usually the mean turbidity values obtained from replicate tubes and the assay may be conducted using a Latin square arrangement of tubes incubated in a shaker, which is necessary to ensure adequate aeration and uniform growth throughout the tube.
Practical aspects of the conduct of turbidimetric assays.
Incubation time is critical in two respects. First, it is necessary to ensure that the culture in each of the many tubes in the incubator has exactly the same incubation period, because errors of a few minutes become significant in a total of only 3–4 hours’ incubation. Care must therefore be taken to ensure that the tubes are inoculated in a precise order, and that growth is stopped in the same order by the addition of formalin, heating or other means.
The incubation period must be appropriate to the inoculum level so that the cultures do not achieve maximal growth. At the concentrations used for such assays, the antibiotics usually reduce growth rate but do not limit total growth. Therefore, if the incubation period is sufficiently long, all the cultures may achieve the same cell density regardless of the antibiotic concentration.
There are certain other limitations to the use of turbidimetric assays. Because it is the ‘cloudiness’ of the culture that is measured, standard and test solutions in which the organisms are suspended should, ideally, be clear before inoculation. Cloudy or hazy solutions which may result from the extraction of the antibiotic from a cream, for example, can only be determined after similarly compensating the standards or otherwise eliminating the error. Test organisms that produce pigments during the course of the incubation period should be avoided; so too should those that normally clump in suspension.
The rate of growth of the test organism may vary significantly from one batch of medium to another. Thus it is important to ensure that all the tubes in the assay contain medium from the same batch, and were prepared and sterilized at the same time. Many liquid media become darker brown on prolonged heating, and so samples from the same batch may differ in colour if the sterilizing time is not strictly controlled.
Minimum inhibitory concentration determinations (MICs)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


