Peripheral T-Cell Lymphoma, Unspecified
Definition
Peripheral T-cell lymphomas (PTCL) are neoplasms of mature T-cell lineage that usually present with a lymphomatous clinical picture. In the World Health Organization (WHO) classification, the term unspecified (U) is an optional addition to emphasize that these neoplasms do not belong to any of the other more specifically defined T-cell lymphomas or leukemias (1). The terms not otherwise specified or not otherwise categorized also have been used in lieu of “unspecified” in the literature.
The category of PTCL-U is thought to be a heterogeneous group composed of more than one disease that currently cannot be further subdivided based on current knowledge and diagnostic tools. As a result, the category of PTCL-U is fluid, and its composition will likely change over time. When a group of lesions within the category of PTCL-U is recognized to have specific clinicopathologic or molecular features, this group will most likely be reclassified to another, more specific entity. Hepatosplenic T-cell lymphoma is an example of a group of neoplasms that once would have been designated as PTCL-U, but is now designated separately in the WHO classification based on its distinctive clinicopathologic features (2). Conversely, lesions may be designated as a distinct entity and, following improved understanding and redefinition, a subset of tumors may return to the PTCL-U category. ALK–anaplastic large-cell lymphoma is a potential example in the latter category as will be discussed (3).
Synonyms
Immunoblastic sarcoma of T cells (4); lymph node–based T-cell lymphoma (5); the following T-cell lymphoma types in the Kiel classification (6): pleomorphic small-cell lymphoepithelioid (Lennert), T zone, pleomorphic medium- and large-cell, and immunoblastic; post-thymic T-cell lymphoma; mature T-cell lymphoma.
Epidemiology and Pathogenesis
T/NK-cell lymphomas and leukemias have been reported to account for approximately 12% to 15% of non-Hodgkin lymphomas in the Western hemisphere (7,8). In the United States, however, the frequency of T/NK-cell lymphomas appears to be lower. According to the Surveillance, Epidemiology, and End Results (SEER) registry from 1992–2001, 5.4% of all lymphoid tumors were T/NK-cell neoplasms (9). The incidence rate for all T/NK-cell neoplasms was 1.79 per 100,000 person-years (9). These neoplasms are more common in males than females, and the male-to-female ratio ranges from 1.5 (African-Americans) to 1.9 (whites). T/NK-cell neoplasms are slightly more frequent in African-Americans than whites. In Asians, T/NK-cell neoplasms are relatively more frequent, but their absolute incidence is similar to that of whites and African-Americans. The overall incidence of T/NK-cell neoplasms in the SEER registry increased approximately 4% per year, from 2.0 to 2.6 per 100,000 person-years in males and 1.0 to 1.4 person-years in females. The incidence of T/NK-cell neoplasms increases markedly with age (9).
In the SEER registry, unspecified T/NK-cell neoplasms and PTCL-U are reported separately, which complicates assessment. However, the most common type of T-cell lymphoma is PTCL-U. In the Non-Hodgkin Lymphoma Classification Project, PTCL-U represented 6.0% of all non-Hodgkin lymphomas and approximately half of all T/NK-cell neoplasms (7). The epidemiologic features of PTCL-U closely follow that for all T/NK-cell neoplasms in the SEER registry. The incidence of PTCL-U increases with age, and these lymphomas are more common in men than in women, and in African-Americans more than in whites.
Very little is known about the etiology or pathogenesis of PTCL-U. This is, in part, definitional. Once the etiology or pathogenesis of a group of neoplasms is understood, this group is likely to be classified separately.
Clinical Findings
The median age of patients with PTCL-U has ranged from 51 to 68 years in various studies (10,11,12,13,14,15,16). Systemic (B) symptoms are common, present in 44% to 57% of patients, and approximately 75% of patients have clinical stage III or IV disease. Lymph nodes are the main site of PTCL-U, but extranodal sites are frequently involved, in 36% to 79% of patients in various studies. Extranodal sites commonly involved include the bone marrow, spleen, liver, lung, and skin. Bone marrow involvement has ranged from 14% to 50% in various studies (10,11,12,13,14,15). Many patients have a poor performance status. An elevated serum lactate dehydrogenase (LDH) level is common, in over half of all patients in different studies (11,12,14,15,16). Bulky disease can occur in approximately 10% of patients (10,12,14). Many patients with PTCL-U have a high International Prognostic Index (IPI). Patients with PTCL-U rarely present with a clinical picture of leukemia, but low-level blood involvement can be detected in a small subset of patients. Patients with PTCL-U can have cytokine-related paraneoplastic phenomena including pruritus, eosinophilia, and hemophagocytic syndrome (1). In some patients, PTCL-U can be preceded by an immunologic disorder, such as Hashimoto thyroiditis, immune thrombocytopenic purpura, and rheumatoid arthritis (17).
The prognosis of patients with PTCL-U is poor. The 5–year overall survival has ranged from 25% to 45% in various studies (10,13,14). In the study by Gallamini and colleagues (14), factors that correlated with poorer prognosis included older age (>60 years), low performance status, high serum LDH level, and bone marrow involvement. Other studies have correlated poor prognosis with advanced stage of disease and
high IPI (10,11,13,15). However, the small subset of patients who present with localized disease and a low IPI have a better outcome. T-cell lineage, by itself, imparts a poorer prognosis (1,16).
high IPI (10,11,13,15). However, the small subset of patients who present with localized disease and a low IPI have a better outcome. T-cell lineage, by itself, imparts a poorer prognosis (1,16).
Histologic Findings
In most biopsy specimens, PTCL-U completely replaces the lymph node architecture (1,12,13,15,16,17,18,19). In some cases, the neoplastic cells are associated with fibrosis, and fibrous bands can irregularly compartmentalize the neoplasm simulating a nodular pattern. In a subset of biopsy specimens, the lymph node architecture is partially replaced, and PTCL-U preferentially involves the paracortex or T-zone areas. A subset of PTCL-U can be associated with a proliferation of postcapillary venules in a fine, interweaving (arborizing) fashion.
Cytologically, PTCL-U is exceptionally heterogeneous, attributable to both the neoplastic cells and associated reactive cells typically present in PTCL-U (1,18,19). The neoplastic T-cells can exhibit a wide cytologic spectrum, ranging from small to intermediate-sized to large; one of these cell types can predominate, or a mixture of these cells can be present (Figs. 73.1,73.2,73.3). Of these, the small-cell type of PTCL-U is the least frequent; most cases are composed of intermediate-sized or large cells. The neoplastic cells can have sparse or abundant cytoplasm that can be clear, eosinophilic, or basophilic. The nuclei of the neoplastic cells can be vesicular or hyperchromatic. Nuclear pleomorphism is common, and bizarrely shaped or multinucleated nuclei can occur (Fig. 73.4). Reed-Sternberg–like cells can be identified in a subset of cases. The neoplastic cells can show high rates of proliferation and apoptosis (Fig. 73.5). Superimposed on this heterogeneity in the neoplastic cells is the common presence of many reactive cells. These cells are present because neoplastic T cells commonly retain at least some of the functions of normal T cells, including the ability to secrete chemokines and cytokines and to stimulate normal immune cells. The inflammatory cells most commonly identified in PTCL-U include small lymphocytes, epithelioid histiocytes, eosinophils, and plasma cells. Sarcoid-like granulomas also can occur in patients with PTCL-U, either associated with the neoplasm or at other anatomic sites such as skin (20).
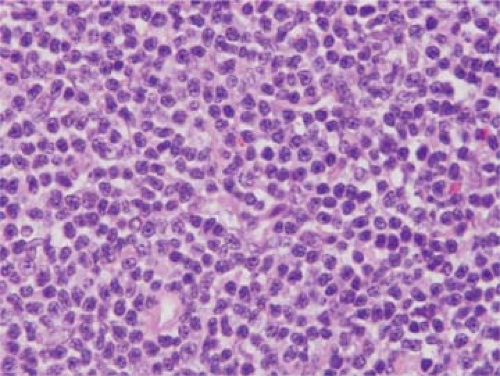 Figure 73.1. Peripheral T-cell lymphoma unspecified (PTCL-U) involving lymph node. In this case, the neoplastic cells are predominantly small. Hematoxylin-eosin stain. |
 Figure 73.2. PTCL-U involving lymph node. In this case, the neoplastic cells are large with eosinophilic cytoplasm. Hematoxylin-eosin stain. |
 Figure 73.3. PTCL-U involving lymph node. In this case, the neoplastic cells are large with basophilic cytoplasm. Hematoxylin-eosin stain. |
 Figure 73.4. PTCL-U involving dermis of skin. In this case, large, multinucleated cells are present. Hematoxylin-eosin stain. |
 Figure 73.5. PTCL-U involving lymph node. A: This neoplasm has many mitotic figures and a high rate of apoptosis. B: The neoplastic cells in this case expressed cytotoxic proteins. TIA-1 is shown. A, hematoxylin-eosin; B, immunohistochemistry with hematoxylin counterstain. |
The diagnosis of PTCL-U by fine needle aspiration is often challenging because these neoplasms can be composed of a spectrum of small, intermediate, and large neoplastic cells that are associated with many inflammatory cells (Fig. 73.6). Immunophenotyping is required to establish T-cell lineage and may show an aberrant T-cell immunophenotype suggestive of lymphoma.
Morphologic Variants of Peripheral T-Cell Lymphoma, Unspecified
Morphologic variants of PTCL-U can be recognized, but they are not designated as separate entities in the WHO classification because evidence is limited that these variants correlate with distinctive clinical or immunophenotypic features or molecular abnormalities (1). The T-zone and lymphoepithelioid cell variants are specifically described in the WHO classification.
In the T-zone variant, the neoplasm is predominantly confined to the interfollicular region of the lymph node (6). Reactive follicles are preserved and can be hyperplastic. The neoplastic cells are usually small or intermediately sized and can have either clear or eosinophilic cytoplasm; nuclear pleomorphism is not prominent (Fig. 73.7). Vascular proliferation and a heterogeneous mixture of reactive cells are commonly associated with the neoplasm in this variant. A subset of these neoplasms histologically overlap, in part, with angioimmunoblastic T-cell lymphoma (AILT) (Chapter 74). A rare group of nodal PTCL-U also has been described in which the neoplasm has a paracortical and nodular pattern of involvement. These neoplasms may represent an early phase of the T-zone variant (21).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


