INTRODUCTION
Pediatric surgical patients are not merely small adults. The surgical care of children differs markedly from that of adults in many respects, including unique physiologic demands that vary according to age and development. The neonate’s physiologic development is closer to that of a fetus, while adolescents are similar to adults, and infants or children have problems unique to their chronologic and developmental age. Infants and children also suffer from congenital abnormalities and diseases not seen in adults, and their management requires an intimate understanding of the relevant embryology and pathogenesis.
NEWBORN CARE
The newborn infant with a surgically correctable lesion often has other disorders that threaten survival. The care of these babies—particularly the premature and small-for-gestational-age babies, has improved with the emergence of the intensive care nursery. Dramatic advances have been made in the technology of infant monitoring and respiratory support. Low-birth-weight infants can now receive ventilatory support from sophisticated infant respirators for prolonged periods in a precisely controlled microenvironment. Surfactant therapy and high-frequency ventilation has allowed a population of extremely premature infants to survive. Temperature is controlled by servoregulation, while pulse and blood pressure are continuously recorded. Ventilation is monitored by transcutaneous O2 and Co2 electrodes or by indwelling arterial catheters. The metabolic consequences of prematurity and intrauterine growth retardation are monitored by frequent measurement of glucose, calcium, electrolytes, and bilirubin in microliter quantities of blood. Nutritional requirements for growth and development can be provided by enteral or parenteral routes. This kind of specialized care of critically ill newborns requires trained personnel and specialized equipment. The care of such babies is best accomplished in designated regional centers capable of providing pediatric surgical and neonatal intensive care.
Newborn infants can be classified according to their level of maturation (weight) and development (gestational age). A normal full-term infant has a gestational age of 37-42 weeks and a body weight greater than 2500 g. The gestational age of the infant is calculated from the date of the last normal menstrual period. However, clinical assessment of gestational age by morphologic and neurologic examination of the small infant can be more accurate than calculation from the menstrual history.
Four signs may be useful in assessing gestational age. Infants less than 37 weeks’ gestational age have (1) fine fuzzy hair with thin, semitransparent skin, (2) ears that lack cartilaginous support, (3) a breast nodule less than 3 mm in diameter, and (4) few transverse creases on the balls of the feet anteriorly. In males, the testicles are incompletely descended and reside in the inguinal canal, and the scrotum is small with few rugae. In females, the labia minora are relatively enlarged and the labia majora are small.
Preterm infants are those born before 37 weeks’ gestation. Several physiologic abnormalities may coexist in preterm infants. Apneic and bradycardic episodes are common and may represent an immature central nervous system (CNS) or, conversely, may represent signs of physiologic instability, most notably with sepsis. The lungs and retinas of preterm infants are very susceptible to high oxygen levels. Retinopathy of prematurity from oxygen toxicity may lead to blindness. Relatively brief exposures to high oxygen concentrations, often coupled with barotrauma from the mechanical ventilator, may damage the lungs, resulting in bronchopulmonary dysplasia. Shunting across a patent ductus arteriosus is common and may lead to pulmonary hemorrhage and congestive heart failure. The preterm infant has a friable choroids plexus and is thus susceptible to intraventricular hemorrhage when stressed in the first week of life. The premature infant may be unable to tolerate oral feeding due to a weak suck reflex. Tube feeds or total parenteral nutrition may be required. Preterm infants have increased requirements for glucose, calcium, and sodium as well as a propensity for hypothermia, impaired bilirubin metabolism, polycythemia, and metabolic acidosis. These problems are accentuated in very low-birth-weight (VLBW) infants or “micropremies” (birth weight < 1000 g).
A small-for-gestational age (SGA) infant is one who is less than the 10th percentile in weight for their gestational age. An SGA infant is the product of a pregnancy complicated by any one of several placental, maternal, or fetal abnormalities. Although body weight is low, their body length and head circumference are age-appropriate. Compared with the premature infant of equivalent weight, the SGA infant is developmentally more mature and faces different physiologic problems. Intrauterine malnutrition results in reduced body fat and decreased glycogen stores. Their relatively large surface area and high metabolic rate predisposes them to hypothermia and hypoglycemia. SGA infants also have an increased risk of meconium aspiration syndrome. Polycythemia (which may lead to complications of hyperviscosity syndrome) is common and necessitates close monitoring of their hematocrit. Due to their relatively mature organ development and function (compared to preterm infants), retinopathy of prematurity, intraventricular hemorrhage, and hyaline membrane disease are uncommon.
Infants and children are susceptible to heat loss because they have a relatively greater body surface area and a thinner subcutaneous fat layer compared with adults. Heat loss occurring by conduction, convection, evaporation, and radiation may be four times that of the adult and is further increased in the preterm infant. Infants are homeotherms and will expend metabolic energy to stay warm at the cost of other functions. Heat is generated not by shivering but by metabolizing brown fat reserves (nonshivering thermogenesis) in response to norepinephrine. This has practical consequences since brown fat may be rendered inactive by some medications (pressors and anesthetic agents) and may be depleted by poor nutrition. Exposure to cold environments increases metabolic work and caloric consumption. Due to limited energy reserves and thin skin, prolonged exposure may rapidly cause hypothermia. Resultant catecholamine secretion increases the metabolic rate (particularly in the myocardium) and produces vasoconstriction with impaired tissue perfusion and increased lactic acid production.
Thus, it is important to maintain the sick newborn in an optimal thermal environment. This is the ambient temperature in which a baby, at a minimal metabolic cost, can maintain a constant and normal body temperature by vasomotor control. To attain such an environment, the gradient between the skin surface and the environmental temperature must be less than 1.5°C. As the skin surface temperature averages 35.5°C, the optimal environmental temperature is 34°C (slightly higher for premature infants). The neonate’s environmental temperature is best controlled by placing the infant in an enclosed incubator. An open radiant warmer is used when the infant is sick and frequent access is necessary. Either the ambient temperature of the incubator can be monitored and maintained at thermoneutrality, or a servo system can be used. The latter regulates the incubator temperature according to the infant’s skin temperature. Heat loss may be further reduced by wrapping the head, extremities, and as much of the trunk as possible in wadding, plastic wrap, plastic sheets, or aluminum foil.
In the operating room, the temperature of the infant must be continuously recorded by placing a thermistor in the rectum or esophagus. Body heat may be conserved by a heating pad, circulated warm air around the child (bear-hugger), infrared lamp, and warm irrigation fluids. The operating room should be prewarmed and the temperature kept at 20-27°C. Wet sponges and drapes exaggerate evaporative heat losses. Plastic drapes contain body heat and keep the skin dry. One of the most effective means of regulating body temperature is to heat and humidify the inhalational anesthetic gases.
Assisted ventilation is often necessary because of underlying disease (eg, persistent fetal circulation and pulmonary hypertension), medications (eg, opioids, PGE2), or physiologic changes imposed by a surgical procedure (eg, closure of an abdominal wall defect or diaphragmatic hernia). At birth, the baby should be warmed, dried, and stimulated. If the baby shows signs of respiratory distress, the pharynx should be aspirated of mucus, amniotic fluid, or meconium. Inadequate respiration should be assisted with positive pressure via mask and escalated to an endotracheal tube as necessary. There is an evolving role for a laryngeal mask airway (LMA) in newborns over 2.5 kg to establish ventilation, especially in cases of difficult airways due to congenital anomalies where intubation may not be possible. The diameter of an endotracheal tube (uncuffed) should approximate that of the 5th digit or the nares, usually between 2.5 and 4 mm. The full-term newborn usually requires a 3.5-mm tube. An orotracheal tube is preferred to a nasotracheal one to minimize trauma and subsequent infection in the nasal passages. The trachea from the glottis to the carina in the newborn is 7.5-cm long, and placement of the tube into the right or left bronchus must be avoided. For infants, optimal tube placement can be estimated as body weight (kg) + 6 as follows: 7 cm from the lips in a 1-kg infant; 8 cm in a 2-kg infant; and 9 cm in a 3-kg infant. Once placed, the endotracheal tube is firmly fixed in place and connected to an infant ventilator. A small air leak between the endotracheal tube and the airway is necessary to minimize laryngeal and tracheal trauma.
Most infant ventilators are time-cycled flow generators capable of delivering both continuous positive airway pressure (CPAP) and intermittent mandatory ventilation (IMV). IMV is a synthesis of simple mechanical ventilation and CPAP breathing that allows the baby to breathe independently between mandatory breaths provided by the ventilator while a continuous positive pressure is maintained on the airway. CPAP breathing helps keep the terminal airways open and is particularly useful when alveolar collapse develops, such as in hyaline membrane disease or with persistent atelectasis.
The gas mixture flowing into the system should be carefully controlled by an air-oxygen mixing device, and the inspired oxygen concentration should be regulated to avoid excessive oxygen delivery. A reasonable goal for arterial Po2 is around 60-80 torr. The gas should be humidified by using a heated nebulizer as nonhumidified circuits can lead to insensible loses. When the arterial Po2 exceeds 80 torr, the inspired oxygen concentration is gradually lowered toward room air; the end-expiratory pressure is incrementally lowered. When Pco2 is less than approximately 45 torr, the IMV rate can be decreased as well. In this way, the baby is gradually weaned from oxygen and mechanical ventilation. Upon removal of the tube, nasal CPAP can be provided, and the inspired oxygen concentration can be increased if necessary.
In severe respiratory compromise (eg, congenital diaphragmatic hernia [CDH], meconium aspiration syndrome), more complex ventilatory strategies are needed. High-frequency ventilation (jet and oscillatory modes) utilizes low tidal volumes at high rates (up to 600 breaths/min) to minimize the deleterious effects of high airway pressure. Inhaled nitric oxide (iNO) can be administered via the ventilatory circuit and may help relax the small airways and pulmonary vasculature. There is a trend toward allowing higher Pco2 levels (permissive hypercarbia) and lower Po2 levels in order to lessen pulmonary trauma from pressure and oxygen. This has been termed “gentle ventilation.” If gentle ventilation, permissive hypercapnia, and the high-frequency modes of ventilation are ineffective, oxygenation and gas exchange can be accomplished using extracorporeal membrane oxygenation (ECMO). This temporary bypass unit oxygenates the blood through an external circuit as the lungs are left to mature or recover from the underlying disease process. The clinical need for ECMO has diminished with the increased widespread use of iNO, surfactant, high-frequency ventilation and the adoption of permissive hypercapnea as a ventilatory strategy.
Effective fluid and electrolyte management involves (1) calculating the fluid and electrolyte requirements for maintaining metabolic functions, (2) replacing losses (evaporative, third space, external), and (3) considering preexisting fluid deficits or excesses. Taking these factors into consideration, a tentative program is devised for fluid and electrolyte administration. The patient’s response is monitored, and the program is adjusted accordingly.
Monitoring fluid status and acid-base balance can be accomplished by both noninvasive and invasive means. Commonly used noninvasive devices include pulse oximetry, urine output, transcutaneous Co2 monitoring, and sphygmomanometry. For critically ill infants, more invasive means are necessary to assess homeostasis. Blood gas analysis via heelstick (capillary), venous catheter, or arterial catheter is frequently employed. Polyvinyl catheters may be placed via an umbilical artery into the aorta, with the tip positioned at the level of T6-T9 or L3-L4 (confirmed radiographically). Indwelling arterial catheters can also be placed in the radial, femoral, or temporal arteries, either percutaneously or by incision. Central venous access may assist in cases where prolonged venous access is needed or parenteral nutrition is necessary or when blood is frequently sampled. It may be obtained via the umbilical vein; a percutaneously inserted central catheter (PICC) via the saphenous, cephalic, median basilic, or temporal veins; or using a Broviac catheter via the femoral, internal jugular, facial, or subclavian veins.
In the newborn infant, the basic maintenance requirement of water is the volume required for growth and replacement of losses from the skin, lungs, and stool. Requirements during the first day of life are unique because of the greatly expanded extracellular fluid volume in the newborn baby, which decreases after 24 hours. For example, infants born with intestinal obstruction (eg, intestinal atresia) are initially not hypovolemic as a result of fluid adjustments across the placenta. Up to 10% of a newborn infant’s birth weight is lost in the first 3-7 days; the majority is water loss, with minor contributions from meconium and urine. During the first 24 hours of life, basic maintenance fluid should range from 60 to 80 mL/kg/d for term infants, and from 80 to 100 mL/kg/d for preterm infants. This requirement gradually increases to a minimum 80-100 mL/kg/d by 4 days of life in normal infants. For children and adolescents, the most commonly used method of calculating fluid requirements is based on body weight (Table 43–1). However, because of the many factors affecting maintenance requirements, there is no close or constant relationship between body weight and fluid and electrolyte needs.
In the surgical patient, fluid, serum electrolyte, and acid-base abnormalities are corrected before operation, when feasible. Intraoperative fluid requirements consist of the estimated maintenance requirement plus replacement of preexisting deficits (if uncorrected) plus replacement of intraoperative losses, including blood.
Postoperatively, losses from intestinal drainage and fistulas are directly measured and replaced with an appropriate electrolyte solution (Table 43–2). In neonates, it is wise to measure the electrolytes in the fluid to more accurately guide replacement, especially for proximal intestinal stomas or fistulas. Protein-rich losses (eg, chest tube drainage of a chylothorax) can be replaced with colloid such as an albumin solution or fresh frozen plasma (FFP). Internal losses into body cavities or tissues (third space losses) cannot be measured; adequate replacement of these losses depends on careful monitoring of the patient’s vital signs and urine output. Following an operation such as a laparotomy or thoracotomy, the fluid requirement may exceed 150 mL/kg/d for several days postoperatively. Isotonic fluids are better in the immediate postoperative period in patients over 6 months of age when significant third space losses are possible.
| Electrolyte Content | |||||
|---|---|---|---|---|---|
| Type of Fluid | Na+ (mEq/L) | K+ (mEq/L) | CI2 (mEq/L) | HCO32 (mEq/L) | Replacement |
| Gastric (vomiting) | 50(20-90) | 10(4-15) | 90(50-150) | … | 5% dextrose in half-normal (0.45%) saline plus KCl 20-40 mEq/L |
| Small bowel (ileostomy) | 110(70-140) | 5(3-10) | 100(70-130) | 20(10-40) | Lactated Ringer |
| Diarrhea | 80(10-140) | 25(10-60) | 90(20-120) | 40(30-50) | Lactated Ringer with or without HCO3– |
| Bile | 145(130-160) | 5(4-7) | 100(80-120) | 40(30-50) | Lactated Ringer with or without HCO3– |
| Pancreatic | 140(130-150) | 5(4-7) | 80(60-100) | 80(60-110) | Lactated Ringer with or without HCO3– |
| Sweat | |||||
| Normal | 20(10-30) | 4(3-10) | 20(10-40) | … | … |
| Cystic fibrosis | 90(50-130) | 15(5-25) | 90(60-120) | … | … |
Basic electrolyte and energy requirements are provided by sodium, 3-4 mEq/kg/d (up to 5 mEq/kg/d for preterm infants) in 5% or 10% dextrose, with the addition of potassium, 2-3 mEq/kg/d, once urine production has been established. Calcium gluconate (200-400 mg/kg/d) may be added, especially in preterm infants. Additional electrolytes such as bicarbonate and magnesium are added, as needed.
Many stressed newborn infants develop low blood levels of potassium, calcium, magnesium, and glucose. A deficiency of any one of these will produce such signs as vomiting, abdominal distention, poor feeding, apneic spells, cyanosis, lethargy, eye rolling, high-pitched cry, tremors, or convulsions. Convulsions and tetany due to hypocalcemia should be treated with intravenous 10% calcium solution given at a rate of 1 mL/min while the EKG is carefully monitored. Although hypocalcemia can be largely eliminated by adding calcium salts to intravenous solutions, caution is required since subcutaneous infiltration may produce severe vasoconstriction and skin necrosis. If there is no response to correction of a documented calcium deficiency, hypomagnesemia should be suspected and a serum magnesium level obtained.
Rapid determination of the blood glucose level can be done in the neonatal unit with blood glucose reagent strips. This may be correlated at intervals with serum glucose determinations, the frequency depending on the stability of the infant. Generally, intravenous fluids should contain a minimum of 10% dextrose, and if non–dextrose-containing solutions such as blood or plasma are being administered, close monitoring of the blood glucose level is essential. The treatment of hypoglycemia consists of giving 50% glucose, 1-2 mL/kg intravenously, followed by a continuous infusion of 10%-15% glucose solutions at a rate equivalent to that needed for maintenance water requirements.
Newborns require a relatively large caloric intake because of their high basal metabolic rate, caloric requirements for growth and development, energy needs to maintain body heat, and limited energy reserve. An infant requires calories at a rate of 100-130 kcal/kg/d and protein at a rate of 2-4 g/kg/d to achieve a normal weight gain of 10-15 g/kg/d (Table 43–3). Thirty percent to 40% of the total nonprotein calories should be provided as fat. These requirements decline with age but increase with surgery, sepsis, and trauma or burns. Caloric requirements are increased 10%-25% by surgery, more than 50% by infection, and 100% by burns.
The best means of providing calories and protein is through the gastrointestinal (GI) tract. If the GI tract is functional, standard infant formulas, blenderized meals, or prepared elemental diets can be given by mouth, through nasogastric or nasojejunal feeding tubes, or through gastrostomy or jejunostomy tubes placed surgically. Gastric feeding is preferable because it allows for normal digestive processes and hormonal responses, a greater tolerance for larger osmotic loads, and a lower incidence of dumping. The use of nasoduodenal or nasojejunal tubes is reserved for infants who cannot tolerate intragastric feeding (eg, delayed gastric emptying, gastroesophageal reflux [GER], depressed gag reflex).
The availability of nutritionally complete liquid diets of low viscosity allows continuous feeding through small-diameter catheters. Elemental diets made by mixing crystalline amino acids, oligosaccharides, and fats can be completely absorbed in the small intestine with little residue. Their use is limited because they cause diarrhea as a result of the high osmolality of full-strength formulas. This can be avoided by administering dilute solutions by continuous drip. Initially, the volume of dilute solution is gradually increased, and the concentration is then progressively increased in a stepwise fashion—ie, half strength, three-fourths strength, and full strength. Formulas that remain below 500 mOsm are best.
Small Silastic or polyethylene catheters such as those used for intravenous infusion can be passed through the nose or mouth into the stomach or jejunum. In more complex cases, a surgically placed gastrostomy or jejunostomy may be necessary for postoperative feeding. A variety of techniques and methods are employed in their construction. In the case of a gastrostomy, either a balloon catheter is used (ie, Foley) or a low-profile gastrostomy button is placed. Silastic is superior to other plastics because it does not become rigid when exposed to intestinal contents. Parenteral nutrition combined with enteral feeding is often necessary for infants with short bowel syndrome until intestinal adaptation occurs.
The indications for parenteral alimentation include the following: (1) expected period of prolonged ileus (eg, following repair of gastroschisis or high jejunal atresia); (2) intestinal fistulas; (3) supplementation of oral feedings, as in intractable diarrhea, short bowel syndrome, or various malabsorption syndromes; (4) intrauterine growth retardation; (5) catabolic wasting states such as infections or tumors when gastric feedings are inadequate or not tolerated; (6) inflammatory bowel disease; (7) severe acute alimentary disorders (pancreatitis, necrotizing enterocolitis [NEC]); and (8) chylothorax.
Concentrated solutions (12.5% glucose or more) thrombose peripheral vessels. Placement of a central venous catheter (PICC or Broviac) into the superior or inferior vena cavae allows the large blood flow to dilute the solution immediately, allowing more concentrated sugar solutions (15%-30% glucose) to be administered. The catheter may be placed percutaneously through the subclavian or internal jugular vein or inserted by cut down into the external jugular, anterior facial, internal jugular, cephalic, brachial, or saphenous veins. For long-term use, Broviac (single lumen) or Hickman (double lumen) catheters, with Dacron cuffs positioned near the exit site of the skin, are preferred to minimize infection and to prevent accidental dislodgment.
Intravenous alimentation solutions containing an amino acid source (2%-5% crystalline amino acids or protein hydrolysate), glucose (10%-40%), electrolytes, vitamins, and trace minerals are used. The electrolyte composition of the protein solution should be known so that the desired composition of the final solution can be adjusted by appropriate additives according to the individual patient’s requirements. A standard solution suitable for infants and young children must contain calcium, magnesium, and phosphate to allow for growth. Trace minerals are also added to the basic solution (Table 43–4). These solutions should be infused at a constant rate with an infusion pump to avoid blood backing up the catheter and clotting and to prevent wide fluctuations of blood glucose and amino acid concentrations. If it is necessary to restrict the volume of infusion, more concentrated glucose solutions can be used to increase the caloric intake.
| Component | Neonate | 6 mo to 10 y | >10 y |
|---|---|---|---|
| Calories (kcal/kg/d) | 90-120 | 60-105 | 40-75 |
| Fluid (mL/kg/d) | 120-180 | 120-150 | 50-75 |
| Dextrose (mg/kg/min) | 4-6 | 7-8 | 7-8 |
| Protein (g/kg/d) | 2-3 | 1.5-2.5 | 0.8-2.0 |
| Fat (g/kg/d) | 0.5-3.0 | 1.0-4.0 | 1.0-4.0 |
| Sodium (mEq/kg/d) | 3-4 | 3-4 | 3-4 |
| Potassium (mEq/kg/d) | 2-3 | 2-3 | 1-2 |
| Calcium (mg/kg/d) | 80-120 | 40-80 | 40-60 |
| Phosphate (mg/kg/d) | 25-40 | 25-40 | 25-40 |
| Magnesium (mEq/kg/d) | 0.25-1.0 | 0.5 | 0.5 |
| Zinc (μg/kg/d) | 300 | 100 | 3 mg/d |
| Copper (μg/kg/d) | 20 | 20 | 1.2 mg/d |
| Chromium (μg/kg/d) | 0.2 | 0.2 | 12 mg/d |
| Manganese (μg/kg/d) | 6 | 6 | 0.3 mg/d |
| Selenium (mg/kg/d) | 2 | 2 | 10-20 mg/d |
Complications of prolonged intravenous alimentation are numerous. The most frequent problem is catheter sepsis. Although catheter removal will quickly treat the problem, a trial of antibiotics effective against gram-positive and gram-negative pathogens is indicated. Catheter removal is indicated in the presence of worsening sepsis, three positive blood cultures, or documented yeast infection (with antifungal treatment after catheter removal). Clotting in the catheter may be controlled by adding 1 unit of heparin per milliliter of solution. Emphasis on a constant rate of infusion will minimize hyperglycemia or hypoglycemia. Analysis of serum electrolytes (including calcium and phosphate) may be necessary several times a week initially, but the interval is decreased to once a week when the patient is stable. Patients must be observed for hyperammonemia and for vitamin or trace mineral deficiency. Progressive hepatomegaly and jaundice of uncertain origin can occur after prolonged parenteral alimentation. This syndrome may subside when the parenteral solution is discontinued or when it is infused for a period of 12-16 hours and then the infusion is stopped for 8-12 hours (cycling) or when augmented with enteral feeding. Use of an ω-3 fatty acid based emulsion (Omegaven) has shown potential in moderating the effects of TPN cholestasis.
Total blood, plasma, and red blood cell (RBC) volumes are higher during the first few postnatal hours than at any other time in an individual’s life. Several hours after birth, plasma shifts out of the circulation, and total blood and plasma volume decrease. The high RBC volume persists, decreasing slowly to reach adult levels by the seventh postnatal week. Age-related estimations of blood volume are summarized in Table 43–5.
Although not clinically significant, both the prothrombin time and the partial thromboplastin time may be slightly prolonged at birth due to relative deficiencies of clotting factors. Defects in the coagulating mechanism may occur in newborn infants as a result of vitamin K deficiency, thrombocytopenia, inherited disorders, and temporary hepatic insufficiency due to immaturity, asphyxia, or infection. It is standard to administer 1.0 mg of vitamin K intramuscularly to all newborns.
The blood lost during operation varies greatly according to the complexity of the operative procedure, the underlying disease, and the effectiveness of hemostasis. Mild blood loss, amounting to less than 10% of the blood volume, usually does not require transfusion. It is imperative to develop methods for closely monitoring the amount of blood lost during operations since significant blood loss is often underestimated in the newborn, especially the preterm infant. Dry sponges should be used and weighed shortly after use to minimize error from evaporation. The suction line, connected to a calibrated trap on the operating table, should be short to diminish the dead space of the tubing and to provide immediate data about accumulated blood loss. Visual observation may be used as a rough guide, but it tends to give a falsely low estimate of the loss.
Before operation, newborn infants should receive vitamin K, 1.0 mg intravenously or intramuscularly. If an extensive surgical procedure is anticipated, the patient’s blood should be typed and crossmatched in case transfusion is required. In infants with hematocrits greater than 50%, blood loss may be replaced by infusing lactated Ringer’s solution or FFP to compensate for losses of up to 25% of total blood volume. Greater blood losses should be replaced with packed RBCs. A transfusion of RBCs at a volume of 10 mL/kg should raise the hematocrit 3%-4%. The transfused blood should be prewarmed to body temperature by running it through coiled tubing immersed in water at 37°C. With excessive blood loss, clotting factors and platelets can be depleted rapidly, and FFP and platelets of identical blood type should be available. With massive transfusion a ratio of 1:1 of FFP to RBCs and 1:2 of platelets to RBCs has been shown to improve outcomes.
The importance of gastric decompression in the surgical newborn cannot be overemphasized. The distended stomach carries the risk of aspiration and pneumonia and may also impair diaphragmatic excursion, resulting in respiratory distress. For example, oxygenation and ventilation in a neonate with CDH may become progressively impaired as the herniated intestine becomes distended with air and fluid. With gastroschisis, omphalocele, and diaphragmatic hernia, the ability to reduce the prolapsed intestine into the abdominal cavity is impaired by intestinal distention. It is critical to avoid bag-mask ventilation in these patients. A double-lumen (sump) tube, such as a 10F Replogle or Anderson tube, is preferred, utilizing low continuous suction. If a single-lumen tube is used, intermittent aspiration by syringe or machine is required. The correct position of the tube in the stomach is confirmed by carefully measuring the tube prior to insertion and by radiographs. Careful taping of the tube is essential to avoid displacement.
Blood analyses should be restricted to those studies essential for diagnosis and management. The volume of blood drawn for laboratory tests should be documented as these small volumes cumulatively represent significant blood loss in a small infant. Generally, the only “routine” preoperative blood analyses for a neonate consist of a complete blood count and a blood specimen for type and crossmatch (in the case of major newborn surgery). Electrolytes in the first 12 hours of life simply reflect the mother’s electrolytes. Coagulation studies (eg, PT, PTT, ACT) are rarely indicated.
The following are general guidelines, but institutional practices vary considerably. The guiding principles in setting a standard include: risk of hypoglycemia associated with NPO status, tolerance or comfort level of the NPO child, and the desire to have an empty stomach upon induction of general anesthesia to mitigate aspiration risk.
No solids, breast milk, or formula 4 hours prior to the procedure. Infants may have clear liquids (water, oral electrolyte mixtures, glucose water, or apple juice) until 2 hours prior to the procedure.
Nothing to eat or drink after midnight except clear liquids (water, apple juice, oral electrolyte mixtures, gelatin dessert, white grape juice), which can be continued until 2 hours prior to the procedure.
Nothing to eat or drink after midnight except clear liquids (water, apple juice, plain gelatin desserts) until 4-6 hours prior to the procedure.
The bowel is mechanically cleansed for elective bowel resection. Opinion varies about whether a bowel preparation is needed for certain procedures as well as about what to use to accomplish it and whether to do it at home or in the hospital. An inpatient regimen begins the day prior to surgery and consists of polyethylene glycol-electrolyte solution (GoLYTELY), 25 mL/kg/h for 4 hours or until the effluent is clear. Metoclopramide (0.1 mg/dose IV) is given 1 hour before the GoLYTELY. Pedialyte can be given ad lib until the time to have nothing by mouth.
Outpatient preparations are reserved for patients over 1 year of age. Clear liquids are given the day before surgery. Bisacodyl (Dulcolax) suppositories and 8-oz lukewarm tap water enemas can be given the morning and in the evening the day before surgery. For children over 5 years of age, magnesium citrate is added (1 oz per year of age up to a maximum of 8 oz) and given orally in the morning and evening the day before surgery, along with 16-oz tap water enemas.
LESIONS OF THE HEAD & NECK
Dermoid cysts are congenital inclusions of skin and skin appendages commonly found on the scalp and eyebrows and in the midline of the nose, neck, and upper chest. They present as painless swellings that may be completely mobile or fixed to the skin and deeper structures. Dermoid cysts of the eyebrows and scalp may produce a depression in the underlying bone that appears as a smooth, punched-out defect on radiographs of the outer table of the skull. They do not extend intracranially. In contrast, cysts of the face and scalp that are located in the midline may represent an alternative diagnosis of encephalocele and would be handled much differently so it is imperative to obtain an MRI or CT scan preoperatively. Dermoid cysts of the midline neck may be confused with thyroglossal duct cysts. However, dermoids do not move with swallowing or protrusion of the tongue since they are not deep to the strap muscles, unlike thyroglossal cysts. All dermoids contain a cheesy material that is produced by desquamation of the cells of the epithelial lining. Care should be taken when planning the operative approach to facial dermoids to avoid either incomplete excision or unnecessary surgical scarring in cosmetically sensitive areas. Dermoids should be excised intact, since incomplete removal will result in recurrence. Those lesions arising near the eyebrows should be excised through an incision adjacent to the hairline. The eyebrows should not be shaved nor should the incision go through any eyebrow follicles since a permanent glabrous area will develop. Recently, tunneled endoscopic approaches originating from behind the hairline have been used successfully to avoid facial scarring.
During the first month of fetal life, the primitive neck develops four external clefts and four pharyngeal pouches that are separated by a membrane. Between the clefts and pouches are branchial arches. The dorsal portion of the first cleft becomes the external auditory canal; the other clefts are obliterated. The pharyngeal pouches persist as adult organs. The first pouch becomes the auditory tube, the middle ear cavity, and the mastoid air cells. The second pouch incompletely regresses and becomes the palatine tonsil and the supratonsillar fossa. The third pouch forms the inferior parathyroid glands and thymus; the fourth forms the superior parathyroid glands. Branchial anomalies are remnants of this fetal branchial apparatus.
A tract of branchial origin may form a complete fistula, or one end may be obliterated to form an external or internal sinus, or both ends may resorb, leaving an aggregate of cells forming a cyst (Figure 43–1). Fistulas that arise above the hyoid bone and communicate with the external auditory canal represent persistence of the first branchial cleft. These tracts are always lined by squamous epithelium. Cysts and sinuses of second or third branchial origin are lined by squamous, cuboidal, or ciliated columnar epithelium. Fistulas that communicate between the anterior border of the sternocleidomastoid muscle and the tonsillar fossa are of second branchial origin, and those that extend into the piriform sinus are derived from the third branchial pouch. Cysts developing from branchial structures usually appear later in childhood as opposed to sinuses and fistulas. Branchiogenic anomalies occur with equal frequency on each side of the neck, and 15% are bilateral. Second branchial cleft abnormalities are most common, occurring six times more frequently than first cleft anomalies.
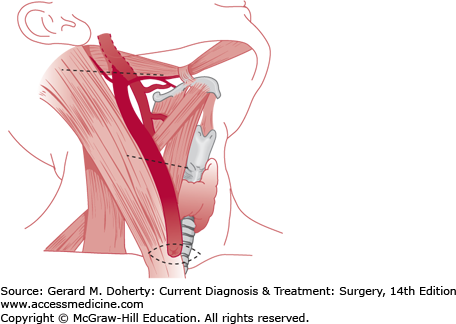
A sinus or fistulous opening along the anterior border of the sternocleidomastoid muscle may be noted at birth and usually discharges a mucoid or purulent material. The patient may complain of a foul-tasting discharge in the mouth upon massaging the tract, but the internal orifice is rarely recognized. Some may present with an acute infection. The cysts are characteristically found anterior and deep to the upper third of the sternocleidomastoid muscle, or they may be located within the parotid gland or pharyngeal wall, over the manubrium, or in the mediastinum. Sinuses and cysts are prone to become repeatedly infected, producing cellulitis and abscess formation. Incomplete branchial sinuses appear as a dimple that contain cartilage and do not drain or communicate with the deep structures of the neck.
Granulomatous lymphadenitis due to mycobacterial infections may produce cystic lymph nodes and draining sinuses, but these are usually distinguishable by the chronic inflammatory reaction that precedes the purulent discharge. Suppurative lymphadenitis, most commonly due to Staphylococcus aureus, may resemble an infected branchial remnant. However, treatment and complete healing of the lymphadenitis is curative, whereas an identifiable branchial remnant will persist after the infection resolves. Hemangiomas and lymphatic malformations (LM) are soft, spongy tumor masses that might be confused with branchial cysts, but the latter have a firmer consistency. LM may transilluminate, while branchial cysts do not. Carotid body tumors are quite firm, are located at the carotid bifurcation, and occur in older patients. Lymphomas produce firm masses in the area where branchial remnants occur, but multiple matted nodes rather than a solitary cystic tumor distinguish these lesions. Mucoid material may be expressed from the openings of branchial sinuses or fistulas, and a firm cord-like tract may be palpable along its course.
Nearly all branchial abnormalities should be excised early in life since repeated infection is common, making resection more difficult. Asymptomatic, small cartilaginous remnants may be watched, but they are usually removed for cosmetic reasons as well as the smaller risk of infection, compared to the true cyst/fistula. Infected sinuses and cysts require initial incision and drainage. Excision of these tracts is staged and usually performed approximately 6 weeks later, when the acute inflammatory reaction has subsided. Every effort should be made to excise the entire cyst wall or fistula tract (including the skin punctum, if present) since recurrence and infection are common with incomplete removal. Excision should be undertaken cautiously, as the tracts may lie adjacent to the facial, hypoglossal, and glossopharyngeal nerves as well as the carotid artery and internal jugular vein.
Preauricular sinuses, cysts, and cartilaginous rests arise from anomalous development of the auricle and are unrelated to branchial anomalies. The sinuses are often short and end blindly. They can be cosmetically unappealing and often become infected. Superficial skin tags and cartilaginous rests are easily excised without risk to other structures. Preauricular sinus tracts, however, may be very deceptive in their extent, and one should be prepared to proceed with extensive dissection that risks damage to branches of the facial nerve.
LMs are benign multilobular, multinodular cystic masses lined by lymph channel endothelial cells. They result from maldevelopment and obstruction of the lymphatic system. Since they are not proliferative lesions, they should be distinguished from hemangiomas; hence the favored term of lymphatic malformation is currently favored over the more frequently encountered misnomer lymphangioma. Cystic hygroma is another misnomer frequently encountered for cervical lymphatic malformation. Eventually, sequestrations of lymphatic tissue that do not communicate with the normal lymphatic system develop. Fifty percent to 65% appear at birth and 90% by the second year of life. They are located most commonly in the posterior triangle of the neck (75%) (Figure 43–2) and axilla (20%), with the remainder located in the mediastinum, retroperitoneum, pelvis, and groin.
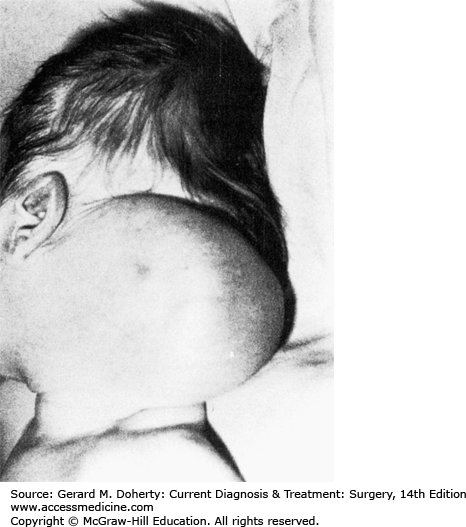
Cervical lymphatic malformations may communicate beneath the clavicle with an axillary hygroma, mediastinal hygroma, or, rarely, both. The majority may be asymptomatic; however, the occult LM usually presents following an upper aerodigestive tract infection as a result of increased or infected lymph flow, or following hemorrhage into the LM from a web of adherent microvasculature. Occasionally, very large lesions occur with involvement of the floor of the mouth, these can cause in utero hydrops or asphyxiation at birth when associated with airway compromise. There is a recognized association between cervical LM and Turner syndrome. These lesions grow along fascial planes and around neurovascular structures; they are infiltrative but not invasive. Large lesions may be recognized prenatally using ultrasound or MRI examination.
There are two modes of treatment, sclerotherapy or excision, the choice of which is based on imaging studies (CT, MRI). Intralesional injection of a sclerosing agent is most effective for unilocular or macrocystic lesions. Examples of agents that have been used are OK-432 (a lyophilized mixture of Streptococcus pyogenes and penicillin G potassium), bleomycin, and doxycycline. Excision is carried out with bipolar cautery to ensure a hemostatic dissection and decrease the incidence of lymph leak and nerve injury. Nevertheless, postoperative lymph leak is common and is treated by closed suction drainage for days to weeks. Intraoperative cyst rupture increases the difficulty of the dissection since the thin-walled cyst is difficult to identify and the margins are obscured. The persistence rate following surgery can be as high as 50% since incomplete excision is the rule rather than the exception in order to avoid potential injury to adjacent neurovascular bundles. Given their infiltrative nature, persistence or symptomatic recurrence following surgical excision and unavoidable surgical scarring, the trend has been increasingly toward sclerotherapy for superficial accessible macrocystic LMs.
During the fourth week of gestation, the thyroid gland develops from an evagination in the floor of the primitive pharynx located between the first pair of pharyngeal pouches. If the anlage of the thyroid does not descend normally, the gland may form at the base of the tongue or remain as a mass anywhere in the midline of the neck along its truncated path of descent. If the thyroglossal duct persists, the epithelial tract forms a cyst that usually communicates with the foramen cecum of the tongue. The thyroglossal duct descends through the second branchial arch anlage, which becomes the hyoid bone, prior to its fusion in the midline. Because of this, the tract of a persistent thyroglossal duct often extends through the hyoid bone (Figure 43–3).
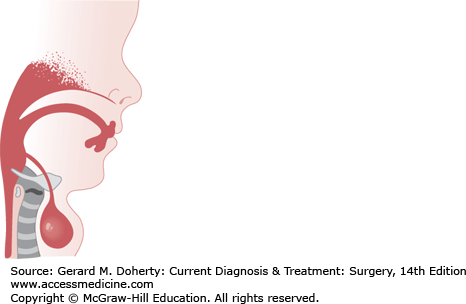
The most common physical finding is a rounded cystic mass of varying size in the midline of the neck just below the hyoid bone. The acute inflammatory reaction of an infection may herald the presence of a cyst. The fluid in the cyst is usually under pressure and may give the impression of being a solid tumor. Cysts and aberrant midline thyroid glands move up and down with swallowing and with protrusion of the tongue since they are deep to the cervical strap muscles. In contrast, lingual thyroid tissue is a rare clinical entity and may produce dysphagia, dysphonia, dyspnea, hemorrhage, or pain.
Lymph nodes, dermoid cysts, and enlarged Delphian nodes containing tumor metastases may be confused with thyroglossal remnants in the midline of the neck. Dermoid cysts do not move with swallowing. Lingual thyroids may be confused with a hypertrophied lingual tonsil or with a ranula, fibroma, angioma, sarcoma, or carcinoma of the tongue. These lesions and thyroglossal cysts may be distinguished from aberrantly located thyroid glands by needle aspiration or by radioiodine scintiscan.
Thyroglossal cysts are prone to infection, and spontaneous drainage or incision and drainage of an abscess will often result in a chronically draining fistula. Excision of an ectopic thyroid may remove all thyroid tissue, producing hypothyroidism. There is a malignant potential of the dysgenetic thyroid tissue located in a thyroglossal duct cyst; carcinoma develops more frequently in ectopic thyroid tissue than in normal thyroid glands.
Complete excision is indicated because of the risk of infection and the possibility of the development of papillary carcinoma later in life. Acute infection in thyroglossal tracts should be treated with antibiotics. Abscesses should be incised and drained. After complete subsidence of the inflammatory reaction (approximately 6 weeks), a thyroglossal cyst and its epithelial tract should be excised. The mid portion of the hyoid bone should be removed en bloc with the thyroglossal tract to the base of the tongue (Sistrunk procedure). Recurrences occur when the hyoid is not removed and when the cyst was previously infected or drained.
Torticollis presents with a hard, nontender, fibrotic mass within the sternocleidomastoid muscle. It may be present at birth but is usually not noticed until the second to sixth weeks of life. The mass appears with equal frequency in both sexes and on each side of the neck. Rarely, there is more than one mass in the muscle or both sternocleidomastoid muscles are involved. A history of breech delivery is present in 20%-30% of these children.
Torticollis is manifested when the sternocleidomastoid muscle is shortened and the mastoid process on the involved side is pulled down toward the clavicle and manubrium. As a result, the head is abducted to the ipsilateral side and rotated to the contralateral side (toward the opposite shoulder). The shoulder on the affected side is raised, and there may be cervical and thoracic scoliosis. Passive rotation of the head to the side of the involved muscle will be resisted and limited to varying degrees, and the muscle will appear as a protuberant band. Because of persistent pressure when the patient is recumbent, the ipsilateral face and contralateral occiput will be flattened. Facial hemihypoplasia and plagiocephaly (flattening of the ipsilateral posterior skull) occurs in untreated cases, usually within 6 months.
Surgery is rarely necessary for this disorder. Torticollis is treated with active range of motion exercises. The child’s shoulders are held flat to a table and the head is tilted and rotated in a full range of motion. This procedure should be performed at least four times a day, usually for 2-3 months. The firm “tumor” often disappears well before the torticollis is cured. If the muscle continues to become progressively shortened, with facial and occipital skull deformity, both heads of the sternocleidomastoid muscle should be divided through a small transverse incision just above the clavicle. This procedure does not reverse the bony changes that have already developed but prevents progression of the process. Recently, endoscopic approaches have been described in order to avoid unsightly surgical scarring in the head and neck region.
CERVICAL LYMPHADENOPATHY
Infections in the upper respiratory passages, scalp, ear, or neck produce varying degrees of secondary lymphadenitis. Most of the causative organisms are streptococcal or staphylococcal species. In infants and young children, the clinical course of the suppurative lymphadenitis may greatly overshadow a seemingly insignificant or inapparent primary infection. Scalp or ear infections produce preauricular or postauricular and suboccipital lymph node involvement; submental, oral, tonsillar, and pharyngeal infections affect the submandibular and deep jugular nodes.
With significant lymphadenitis, the regional lymph nodes become greatly enlarged and produce local pain and tenderness. Enlargement of cervical nodes is most common, followed by occipital and submandibular nodes. Fever is high initially and then becomes intermittent and may persist for days or weeks. The regional nodes may remain enlarged and firm for prolonged periods, or they may suppurate and produce surrounding cellulitis and edema. Subsequently, the nodes may involute or a fluctuant abscess may form, resulting in redness and thinning of the overlying skin. Infected, matted nodes may become so hard as to be indistinguishable (on palpation) from a solid mass.
A smoldering lymphadenitis that neither resolves nor forms an abscess can be confused with granulomatous lymphadenitis, lymphoma, or metastatic tumor. Excisional biopsy is required to differentiate these lesions. After several weeks, there will usually be a reduction in the size and firmness of suppurative adenitis, especially after antibiotic treatment has been started. Recently, methicillin-resistant S. aureus (MRSA) is being encountered at near epidemic levels as a causative agent of suppurative lymphadenitis in the ambulatory setting. A high suspicion of an MRSA infection should be entertained in all children presenting with either a first episode or more certainly in recalcitrant and recurrent cases.
In the acute phase, the patient should be treated with oral or intravenous antistaphylococcal antibiotics. In the subacute or chronic phase, the presence of pus in the node may be confirmed by needle aspiration of the mass. When an abscess is present, it should be incised and drained under general anesthesia. In those cases of MRSA infection, a prolonged course of either vancomycin or linezolid may be required for complete eradication even after drainage.
Although typical tuberculous cervical adenitis is very rare in the United States, atypical mycobacteria (eg, Mycobacterium avium-intracellulare) is encountered and may present as a nonsuppurative area (usually cervical, axillary, or inguinal) of matted nodes with tenderness and a draining sinus. Granulomatous lymphadenitis and caseation may occur in the regional nodes draining the inoculation site of BCG. Cat-scratch disease causes a caseating lymphadenitis in regional lymph nodes (eg, epitrochlear and axillary nodes enlarge after an upper extremity cat scratch).
Children under age 6 years are most frequently affected. The initial manifestation is a painless, progressive enlargement of the lymph nodes in the deep cervical chain and the parotid, suboccipital, submandibular, and supraclavicular nodes. The duration of lymphadenopathy is usually 1-3 months or longer. The nodes may be large and mobile or, with progressive disease, may become matted, fixed, and finally caseate to form an abscess. Incision or spontaneous overlying skin breakdown will result in a chronically draining sinus. In tuberculosis, both sides of the neck or multiple groups of nodes are infected, and the chest radiograph indicates pulmonary involvement. In atypical mycobacterial lymphadenitis, pulmonary disease is rare and the cervical adenitis is unilateral. The tuberculin skin test is weakly positive in over 80% of patients with atypical mycobacterial infection. Skin test antigens from the various strains of atypical mycobacteria are available. A positive skin test helps differentiate granulomatous adenitis from malignant lymphadenopathy. A fluctuant node can be confused with a branchial cleft remnant or a thyroglossal duct cyst.
Cat-scratch disease is usually acquired by a bite or scratch from a kitten. It is caused by a pleomorphic gram-negative bacillus (Bartonella henselae) that is detected in tissues by a silver stain or via serologic testing. It is an acute illness characterized by fever, malaise, possible musculoskeletal manifestations and occasionally a pustular lesion at the site of the scratch. Tender lymph node enlargement usually develops. Two to 4 weeks later, regional lymphadenitis persists, producing painful, fixed suppurative nodes that may develop into a chronically draining sinus.
Atypical tuberculous lymphadenitis may be treated with rifampin (10 mg/kg/d), though definitive treatment usually requires nodal excision. Trimethoprim-sulfamethoxazole may shorten the course of cat-scratch disease and prevent suppuration. When antibiotics are ineffective, the procedure of choice is excision of involved nodes before caseation occurs. Once the nodes become fluctuant or a draining sinus forms, a wedge of involved skin should be excised and the underlying necrotic nodes should be curetted out (rather than excised), taking care not to injure neighboring nerves. The wound edges and skin should be closed primarily. The value of continuing chemotherapy is influenced by sensitivity tests on the cultured material. Excision and primary closure usually result in excellent healing with good cosmetic results.
CONGENITAL CHEST WALL DEFORMITIES
Failure of fusion of the two sternal bars during embryonic development produces congenital sternal cleft, which may involve the upper, lower, or entire sternum. In its severe form, this defect is usually associated with protrusion of the pericardium and heart (ectopia cordis) and congenital heart lesions. Defects may be associated with extracardiac anomalies, including cleft lip, cleft palate, hydrocephalus, and other CNS disorders, or may be one component of the pentalogy of Cantrell. Operative correction is performed in the neonatal period since the chest wall is so pliable; it consists of simple suture approximation of the two sternal halves. More complex defects associated with ectopia cordis are often incompatible with life.
This depression deformity is the most common congenital chest wall abnormality, occurring in one in 300 live births, with a 3:1 male predominance. It is associated with other musculoskeletal disorders (Marfan syndrome, Poland syndrome, scoliosis, clubfoot, syndactyly), and 2% have congenital heart disease. There is a familial form. It results from the unbalanced posterior growth of costal cartilages that are often fused, bizarrely deformed, or rotated. The body of the sternum secondarily exhibits a prominent posterior curvature, usually involving its lower half (Figure 43–4). Commonly, the xiphoid is the deepest portion of the depression. The third, fourth, and fifth costal cartilages are usually affected, though the second to eighth costal cartilages may be involved. The severity of the defect varies greatly from a mild, insignificant depression to an extreme where the xiphoid bone is adjacent to the vertebrae. The depression may be symmetrical or asymmetrical with varying degrees of sternal rotation.
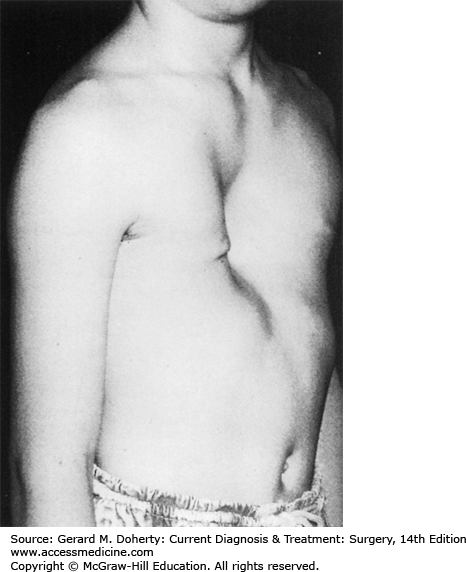
These patients are typically round shouldered, with stooped posture, relative abdominal prominence, flared costal margins, and an asthenic appearance. They may be withdrawn and refuse to participate in sports activities, particularly if their deformity might be exposed. Few patients complain of easy fatigability or inability to compete in exertional activities. Cardiopulmonary function studies rarely demonstrate impairments; this is predominantly a cosmetic deformity with potentially severe psychosocial sequelae.
There is no standard age for repair. Undeniably, it is an easier operation in younger children compared to adolescents. Drawbacks of an early operation include a higher risk of recurrence during the adolescent growth spurt and the inability of a young child to comprehend and assent to a predominantly cosmetic operation. Traditionally, an open repair (Ravitch technique) was performed in which the abnormal cartilages were resected and the sternum was fractured and fixed in a corrected position. Recently, the Ravitch technique has been supplanted by the less invasive Nuss procedure in which a preformed sternal strut is passed, either blindly or with thoracoscopic assistance, under the chest wall muscles, into each hemithorax, and across the mediastinum under the sternum via two small incisions in the midaxillary line. The curved bar is passed upside down and “flipped” into position under the sternum, effectively lifting the sternum and chest wall into a corrected position. The bar is left in place for 2 years, and the patient can resume activity in 3 months. The long-term good to excellent results of the Nuss procedure are better than 95%. The latest evolution in less invasive techniques involves placement of opposing magnetic field implants to draw the chest deformity forward and effect remodeling. In general, there is no cardiopulmonary benefit after chest wall repair except in rare instances when the deformity is excessive. Otherwise, the repair is performed solely to improve appearance. However, the psychosocial benefits of repair of this often embarrassing deformity cannot be minimized.
This is a protrusion deformity, also referred to as pigeon breast or chicken chest. It is approximately ten times less frequent than pectus excavatum. It results from the overgrowth of costal cartilages, with forward buckling and secondary deformation of the sternum (Figure 43–5). Atypical and asymmetric forms with rotation are common. There is a familial form. It is associated with Marfan disease, neurofibromatosis, Poland syndrome, and Morquio disease. Unlike pectus excavatum, the deformity is typically mild or nearly imperceptible in early childhood and becomes increasingly prominent during the rapid growth in early puberty.
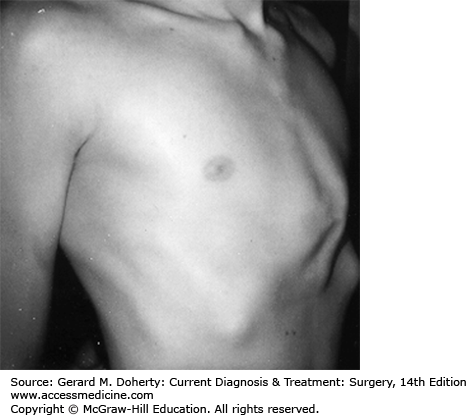
As with pectus excavatum, there is no cardiorespiratory compromise with this deformity, and repair is performed solely to achieve an improved cosmetic appearance. Mild deformities should be left alone and the patient followed to observe for progression. Moderate to severe defects should be repaired, particularly when the patient indicates a desire for improvement. The deformed cartilages are resected, leaving the costochondral membranes (perichondrium) intact. Sternal fracture is usually not necessary. To ensure that the costal cartilages grow back on a straighter line, “reefing” sutures are placed in the perichondrium to shorten them. The costal cartilages regenerate within 6 weeks. A thorough procedure will provide an excellent cosmetic result in nearly all cases. Recurrences are rare. An alternative approach to operative repair is chest bracing via an orthotic vest that needs to fitted and worn by the affected child for several hours daily over several years. This procedure may eventually replace the operative approach, particularly for those patients who are motivated to wear the brace for a majority of each day (about 16 hours).
SURGICAL RESPIRATORY EMERGENCIES IN THE NEWBORN
Certain aspects of respiration unique to the infant must be appreciated. Except during periods of crying, the newborn baby is an obligate nasal breather. The ability to breathe through the mouth may take weeks or months to acquire. Inspiration is accomplished chiefly by diaphragmatic excursion; the intercostal and accessory muscles contribute little to ventilation. Impaired inspiration results in retraction of the sternum, costal margin, and neck fossae; the resulting paradoxical motion may contribute to respiratory insufficiency. The airway is small and flaccid, so that it is readily occluded by mucus or edema, and it collapses readily under slight pressure. Dyspneic infants swallow large volumes of air, and the distended stomach and bowel may further impair diaphragmatic excursion.
Micrognathia—Pierre Robin syndrome
Macroglossia—Muscular hypertrophy, hypothyroidism, lymphatic malformation, Beckwith-Wiedemann syndrome
Anomalous nasopharyngeal passage—Choanal atresia, Treacher-Collins syndrome, Apert syndrome, and Crouzon syndrome
Tumors, cysts, or enlarged thyroid remnants in the pharynx or neck
Laryngeal or tracheal stenosis, webs, cysts, tumors, or vocal cord paralysis
Epiglottitis
Tracheomalacia
Tracheal stenosis with or without complete tracheal rings
Atelectasis
Pneumothorax and pneumomediastinum
Pleural effusion or chylothorax
Pulmonary cysts, sequestration, and tumors
Congenital lobar emphysema
Diaphragmatic hernia or eventration
Esophageal atresia with or without tracheoesophageal fistula
Anomalies of the great vessels (eg, double aortic arch, aberrant left subclavian artery, anomalous origin of left pulmonary artery)
Mediastinal tumors and cysts (foregut duplications, thymomas, substernal goiter, lymphoma)
Pierre Robin syndrome is a congenital defect characterized by micrognathia and glossoptosis, often associated with cleft palate. The small lower jaw and strong sucking action of the infant allow the tongue to be sucked back and occlude the laryngeal airway and may be life threatening.
Infants with mild cases should be kept in the prone position during care and feeding. A nasogastric or gastrostomy tube may be necessary for feeding. Nasohypopharyngeal intubation is effective in preventing occlusion of the larynx. If conservative measures fail, prompt attention to maintaining an open airway by tracheostomy is indicated. Surgical treatment involves tongue placation in which the tongue is sutured forward to the lower jaw, but this frequently breaks down. In time, the lower jaw develops normally. These infants eventually learn how to keep the tongue from occluding the airway.
Complete obstruction at the posterior nares from choanal atresia may be unilateral and relatively asymptomatic. It may be membranous (10%) or bony (90%). When it is bilateral, severe respiratory distress is manifested at birth by marked chest wall retraction on inspiration and a normal cry.
There is arching of the head and neck in an effort to breathe, and the baby is unable to eat. The diagnosis is confirmed by the inability to pass a tube through the nares to the pharynx. With the baby in a supine position, radiopaque material may be instilled into the nares and lateral x-rays of the head taken to outline the obstruction. A CT scan of the nasopharynx will define bony occlusion.
Emergency treatment consists of maintaining an oral airway by placing a nipple, with the tip cut off, in the mouth. The membranous or bony occlusion may then be perforated by direct transpalatal excision, or it may be punctured and enlarged with a Hegar dilator. The newly created opening must be stented with plastic tubing for 5 weeks to prevent stricture.
There are three main types of congenital tracheal stenosis: generalized hypoplasia; funnel-like narrowing, usually tapering to a tight stenosis just above the carina; and segmental stenosis of various lengths that can occur at any level. Tracheomalacia is a functional obstruction in a “soft” trachea that collapses with inspiration. It is often secondary to external compression by vascular anomalies or tumors or from a chronically dilated upper esophageal pouch in those with esophageal atresia.
The diagnostic approach to an infant with respiratory distress and possible distal tracheal obstruction must be carefully integrated with plans for management of the airway, since the compromised infant airway is easily occluded by edema or secretions. This is especially true in distal tracheal lesions, where an endotracheal or tracheostomy tube may not relieve the distal obstruction. The diagnostic value of every procedure must be weighed against the threat of precipitating airway obstruction. Tracheal lesions can be visualized using esophagography, angiography, or CT/MRI scans. Dynamic lesions such as tracheomalacia and vascular compression syndromes are best defined by videotape fluoroscopy or cineradiography with barium in the esophagus. Angiography may be necessary. Flow-volume curves can define the level of obstruction (intrathoracic vs extrathoracic) and the type of obstruction (stenosis vs malacia).
Although bronchoscopy often provides the best delineation of tracheobronchial lesions, it is an invasive procedure that can precipitate acute obstruction from edema or inflammation. A ventilating infant rigid bronchoscope with Hopkins optics should be kept above the critical area to avoid precipitating obstruction. Flexible transnasal awake bronchoscopy is most useful in demonstrating functional abnormalities (eg, malacia).
Noncritical stenotic and malaciac lesions in infants and children should be managed as conservatively as possible, preferably without intubation. “Temporary” stenting of these lesions is seldom temporary, since the presence of the tube itself ensures continued trauma and irritation such that the tube cannot be removed without airway obstruction. If an infant or child cannot be managed without intubation, surgical correction must be considered. Tracheal reconstruction via resection or a variety of tracheoplasty techniques has proved to be the treatment of choice for tracheal lesions. Severe tracheomalacia is treated by addressing the underlying cause. Aortopexy or an endotracheal stent is often necessary. Tracheostomy is a last resort.
CDH is a highly lethal or morbid disease that affects 1 in 2000 live births (Figure 43–6). Anatomically, CDH results from an embryologic fusion defect, allowing herniation of intra-abdominal contents into the chest. Fusion of the transverse septum and pleuroperitoneal folds normally occurs during the eighth week of embryonic development. If diaphragmatic formation is incomplete, the pleuroperitoneal hiatus (foramen of Bochdalek) persists. Intestinal nonrotation is common as the bowel herniates into the thorax rather than undergoing its normal sequence of rotation and fixation. Severe defects cause pulmonary hypoplasia, pulmonary hypertension, and cardiac dysfunction. The larger the hernia and the earlier it occurs, the more severe the pulmonary hypoplasia.
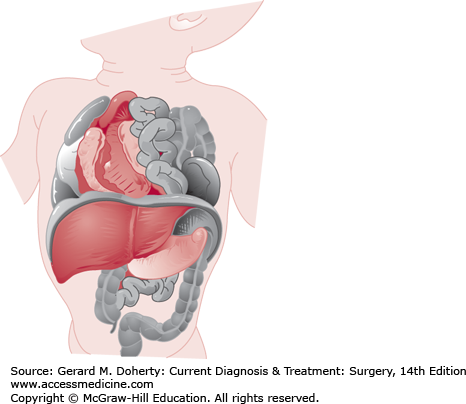
Infants with large diaphragmatic defects are usually symptomatic in the delivery room, with tachypnea, grunting respirations, retractions, and cyanosis, and may require urgent intubation. Smaller defects may not become symptomatic until the infant is several days or months old. Typically, the abdomen is scaphoid since much of the abdominal viscera are in the hemithorax. The chest on the side of the hernia may be dull to percussion, but bowel sounds are not usually appreciated. The left side of the diaphragm is affected four or five times as frequently as the right, with a rate of associated anomalies of 20% (chromosomal abnormalities, neural tube defects, and congenital heart disease). When the hernia is on the left, the heart sounds may be heard best on the right side of the chest.
The development of symptoms with CDH correlates with the degree of pulmonary hypoplasia and pulmonary hypertension. Prenatal diagnosis is occurring more and more frequently and allows the mother and the fetus to be referred to an institution where sophisticated perinatal and pediatric surgical units are available.
A chest radiograph may demonstrate the following: a paucity of gas within the abdomen, radiopaque hemithorax if the bowel does not contain a significant amount of gas or if the left lobe of the liver occupies the majority of the hemithorax, loss of normal ipsilateral diaphragmatic contour, bowel in the thorax, contralateral mediastinal shift, and a coiled nasogastric tube in the hemithorax. Right-sided hernias can be difficult to distinguish from a diaphragmatic eventration. This can be differentiated by an MRI scan. MRI or CT scan can also distinguish between CDH and a cystic lung lesion (eg, congenital cystic adenomatoid malformation).
A nasogastric tube should be placed in the stomach to aspirate swallowed air and to prevent distention of the herniated bowel, which would further compress the lungs. Repair of the diaphragmatic defect is not a surgical emergency, it is rather a physiologic emergency requiring resuscitation, with surgery performed once the infant has stabilized and demonstrated minimal to no pulmonary hypertension. Early (before 48 hours postnatally) hernia repair has been shown to transiently worsen pulmonary function by decreasing pulmonary compliance and increasing airway reactivity. A subcostal abdominal incision should be made and the herniated bowel reduced from the pleural space. Some surgeons prefer a thoracic approach, particularly for right-sided defects. The negative pressure between the bowel and the chest wall may make reduction difficult. Following reduction of the bowel, placement of a chest tube in the pleural space is optional; if used, it is connected to a water seal and not to suction as it may cause physiologically significant mediastinal shift. The diaphragmatic defect should be closed by nonabsorbable sutures. In many instances, a synthetic material is required to close large defects. The abdominal cavity may be too small and underdeveloped to accommodate the intestine and permit closure of the abdominal wall muscle and fascial layers. In such cases, abdominal wall skin flaps should be mobilized and closed over the protruding bowel or a silo created to allow for gradual visceral reduction with concomitant abdominal domain expansion and staged closure of the abdominal wall.
Respiratory support and treatment of hypoxemia, hypercapnia, and acidosis are required before and often after repair. Persistent pulmonary hypertension may result in right-to-left shunt and produce severe hypoxemia in the lower aorta. Nitric oxide added to the ventilation gases can induce pulmonary vasodilation, improve pulmonary perfusion, and reverse the right-to-left shunt. The persistent fetal circulation physiology may be treated successfully in many cases by extracorporeal membrane oxygenation and permissive ventilatory strategies (high-frequency ventilation). Hypoxemic myocardiopathy may require infusion of dopamine to enhance cardiac output. Prenatal treatment for severe CDH (temporary fetal tracheal occlusion to promote lung growth) has been extensively studied and may offer benefit in severe cases of CDH.
The death rate for infants with CDH depends upon the severity of pulmonary hypoplasia, the presence or absence of associated anomalies, and the quality of care provided for these critically ill infants. When diagnosed in utero, prognosis depends on the presence or absence of liver herniation into the left hemithorax, the gestational age at diagnosis, and an ultrasonographic estimation of lung size (the lung-to-head ratio). Long-term, there are a number of measurable physiologic abnormalities that are not necessarily clinically significant such as a reduction in total lung volume, restrictive or obstructive lung disease, and abnormal lung compliance. However, a small subset of patients will survive as “pulmonary cripples” and remain oxygen dependent or ventilator dependent, often requiring tracheostomies. Since there may be deficient periesophageal muscular tissue or an abnormal orientation of the gastroesophageal junction, GER is common. It is most commonly treated nonoperatively, but refractory cases may require a surgical antireflux procedure. Recurrent diaphragmatic hernia occurs in 10%-20% of infants and should be considered in any child with a history of CDH who presents with new GI or pulmonary symptoms. Recurrence is most common when a prosthetic patch is used for the repair.
Surgical units that are immediately adjacent to obstetric services report death rates as high as 80%, because infants with severe pulmonary hypoplasia will be recognized and treated immediately. Infants who survive transfer to surgical centers remote from the delivery area usually have less severe disease, and the death rates reported from these facilities are usually under 40%.
With improvements in prenatal ultrasonographic imaging, many of these defects can be appreciated early enough so that planned delivery at a tertiary facility is possible. Excluding those infants with severe associated anomalies, the overall survival rate using maximal medical therapy has been increasing over the past several years due to advanced ventilation strategies and is well over 70%.
The foramen of Morgagni occurs at the junction of the septum transversum and the anterior thoracic wall. This anterior, central diaphragmatic defect accounts for only 2% of diaphragmatic hernias. It may be parasternal, retrosternal, or bilateral. Unlike Bochdalek hernias, children are typically asymptomatic and the defect is discovered later in life on a chest radiograph taken for reasons unrelated to the hernia. The lateral chest radiograph demonstrating an air-filled mass extending into the anterior mediastinum is pathognomonic. Repair is indicated in the asymptomatic patient due to the risk of bowel obstruction. The viscera are reduced and any associated hernia sac excised. The defect is closed by suturing the posterior rim of diaphragm to the posterior rectus sheath since there is no anterior diaphragm. A prosthetic patch closure is frequently required given the tension that results with native tissue repairs given the absence of anterior diaphragm. Laparoscopic approaches to this type of repair are increasingly being performed. There is no associated pulmonary hypoplasia or hypertension. This defect, when noted in newborns, can be associated with the pentalogy of Cantrell, a disorder with considerable morbidity and mortality that consists of the anterior diaphragmatic defect, distal sternal cleft, epigastric omphalocele, apical pericardial defect, and congenital heart disease (usually a septal defect). Excluding patients with the pentalogy of Cantrell, survival is nearly 100%.
Diaphragmatic eventration is an abnormally elevated or attenuated portion of the diaphragm (or both). It may be congenital (usually idiopathic, but can be associated with congenital myopathies or intrauterine infections) or acquired (as a result of phrenic nerve injury during forceps delivery or surgery). In the congenital form, there is variable thinning or absence of diaphragmatic muscle, at which point its distinction from CDH with a persistent hernia sac is obscure. The elevated hemidiaphragm may produce abnormalities of chest wall mechanics with impaired pulmonary function. Respiratory distress and pneumonia are frequent presenting symptoms, although GI symptoms such as vomiting or gastric volvulus have been reported.
The diagnosis is made by chest radiograph. It is confirmed by fluoroscopy or ultrasound, which demonstrate paradoxical movement of the diaphragm during spontaneous respiration. Incidentally discovered small, localized eventrations do not need repair. Eventrations that are associated with respiratory symptoms should be repaired by plicating the diaphragm using interrupted nonabsorbable sutures.
CONGENITAL LOBAR EMPHYSEMA
Congenital lobar emphysema results from hyperinflation of a single lobe; rarely, more than one lobe is affected. The upper and middle lobes are most frequently involved. Pathologically, there are three forms; hypoplastic emphysema, polyalveolar lobe, and bronchial obstruction.
Hypoplastic emphysema is distinguished by a segment, lobe, or whole lung that has a reduced number of bronchial branches with a diminished number and smaller size of blood vessels. The number of alveoli is abnormally decreased, but the air spaces are too large. The hyperlucent region seen on chest radiograph is normal or small in volume, and since it does not affect the surrounding normal lung, surgical treatment is unnecessary.
Polyalveolar lobe is characterized by a normal size and number of bronchial branches, but there is an abnormal number of alveoli from each respiratory unit. These alveoli are prone to expand excessively, producing emphysema, which encroaches on the surrounding normal lung and therefore requires removal.
Bronchial obstruction may occur from deficient bronchial cartilage support, redundant mucosa, bronchial stenosis, mucous plug, or bronchial compression by anomalous vessels or other mediastinal lesions. With inspiration, the bronchus opens to allow air into the lung, but on expiration the bronchus collapses, trapping the air, and with each respiratory cycle there is progressive expansion of the lobe.
In one-third of patients, respiratory distress is noted at birth; in only 5% of cases do symptoms develop after 6 months. Males are affected twice as frequently as females. The signs include progressive and severe dyspnea, wheezing, grunting, coughing, cyanosis, and difficulty feeding. An increased anteroposterior dimension of the chest and retractions may be seen. The chest is hyperresonant, and decreased breath sounds may be noted over the affected lobe. A chest radiograph may demonstrate radiolucency of the emphysematous lobe, with bronchovascular markings extending to the lung periphery. Compression atelectasis of the adjacent lung, shift of the mediastinum, depression of the diaphragm, and anterior bowing of the sternum can be seen. The emphysematous lobe may continue to expand, compressing adjacent lung and airways, producing progressively severe respiratory distress.
Occasionally, the emphysema may be due to a foreign body or mucous plug in the bronchus that may be aspirated by bronchoscopy. Compression of the bronchus by mediastinal masses may be relieved by removal of the tumor or repair of anomalous vessels.
Treatment of asymptomatic and mildly symptomatic cases may not be necessary. Many patients with lobar emphysema, however, are severely symptomatic, and pulmonary lobectomy is necessary. For those who are breathing spontaneously prior to operation, anesthesia should not be started until all personnel are ready for a rapid thoracotomy since positive-pressure ventilation may acutely enlarge the emphysematous lobe, thereby compressing the normal lung tissue and heart. The prognosis following surgical relief of the lobar emphysema is excellent. Rarely patients may show residual disease in the remaining lung. At long-term follow-up, lung volumes are normal, but the airflow rates are diminished.
GREAT VESSEL ANOMALIES
Tracheobronchial and esophageal compression by the great vessels may occur as a result of anomalies of the aortic arch or of abnormally located or enlarged pulmonary and subclavian arteries. While the most common abnormality is an aberrant right subclavian artery, the most important is the double aortic arch, as it often causes serious respiratory distress in young infants (Figure 43–7). Affected infants have a characteristic inspiratory and expiratory wheeze, stridor, or croup-like cough. Echocardiography, CT, and MRI can demonstrate the anomalous anatomy. Esophagoscopy and bronchoscopy may be helpful in assessing the degree and level of compression. The surgical approach is through the left hemithorax. Following removal of the thymus, the aortic arch and its branches are skeletonized and the anatomy is identified. For the double aortic arch, the smallest arterial component is divided; an anomalous right subclavian artery is divided at its origin. The accompanying fibrous bands and sheaths constricting the trachea and esophagus must also be divided. The ductus arteriosus (or its fibrous remnant) is also divided.
MEDIASTINAL MASSES
Mediastinal masses are relatively common in infants and children and can be classified according to the compartment of the mediastinum from which they arise. The mediastinum is classically divided into anterior, middle, and posterior compartments. Anatomically, the anterior mediastinum consists of the area between the sternum and the anterior aspect of the trachea and pericardium. The middle compartment contains the trachea, major bronchi, and paratracheal spaces. The posterior segment extends from the posterior aspect of the trachea to the spine.
Anterior masses make up one-third and are most commonly teratomas and lymphomas. Teratomas may be cystic or solid and may also be found within the pericardium (middle compartment). Approximately 20% are malignant. Other masses include thymic cysts, thymomas, substernal goiters, and lymphangiomas. Middle mediastinal masses are rare but when present are most likely to be bronchogenic cysts. Sixty percent of mediastinal masses are located within the posterior compartment. Neurogenic tumors are most common and include neuroblastoma, ganglioneuroblastoma, ganglioneuroma, neurofibroma, and neurofibrosarcoma. Common symptoms include respiratory distress (via tracheal or lung compression), Horner syndrome, and pain. Enterogenous cysts or duplications are also commonly seen in the posterior compartment. They are termed neurenteric when there is an associated cervical or thoracic vertebral anomaly.



