CHAPTER 5 Pathology of the marrow
General considerations and infections/reactive conditions
Principles of bone marrow examination
The bone marrow may be examined after aspiration through a special wide-bore needle. The aspirate and trephine biopsy specimens are complementary and when both are obtained, they permit a comprehensive evaluation of the bone marrow. Guidelines based on preferred best practices have been published by the International Council for Standardization in Hematology.1 The usual sites of aspiration are the posterior or anterior iliac crest or manubrium sterni in adults, the posterior superior iliac spine in infants and children and the medial aspect of the upper end of the tibia in neonates. The aspirate, which consists of fragments of marrow tissue, individual nucleated marrow cells and a variable quantity of blood, is spread on glass slides and examined after fixation and staining. Examination of marrow smears allows a detailed analysis of the morphology and cytochemistry of cells and the percentages of various cell types (see Chapter 2 for details). However, differential counts performed on marrow smears do not give accurate data on the prevalence of some cell types, such as megakaryocytes and macrophages, which are relatively resistant to aspiration or have a tendency to remain attached to the aspirated marrow fragments during the preparation of the smears. This drawback may be overcome, to a large extent, by examining preparations obtained by crushing marrow fragments between two glass slides and pulling the slides apart. Apart from its use for cytologic studies with the light microscope, aspirated BM can be used for immunophenotypic, cytogenetic, molecular genetic, biochemical and electron microscope studies.
The histology of the BM is investigated by examining sections of aspirated marrow particles (either in clotted aspirates or after concentration of the particles in various ways) or by examining sections of a core of bone obtained using a specially-constructed trephine needle. The usual site of trephine biopsy is the posterior superior iliac spine. Unlike marrow smears, histologic sections of trephine biopsies permit an appreciation of intercellular relationships and particularly of the relationship between hemopoietic cells, non-hemopoietic cells, the blood vessels and bone (see Chapter 3). Such sections are therefore useful for the detection of granulomas and focal accumulations of malignant cells. They also allow a study of the quantity and distribution of reticulin and collagen in marrow and make possible the diagnosis of myelofibrosis. Histologic sections are usefully stained with hematoxylin and eosin (H&E), Giemsa stain and a reticulin stain. Some laboratories also stain for hemosiderin routinely but an alternative is to apply this stain selectively, only when an aspirate for a Perls stain with an adequate number of fragments is not available.
Alterations of the marrow in disease
Alterations in cellularity
The method of assessing the proportion of the marrow tissue that is composed of hemopoietic cells as opposed to fat cells (percentage cellularity) and the normal values for this parameter are discussed in Chapters 2 and 3. Hypocellularity (i.e. cellularity <25%) is seen in the acquired aplastic or hypoplastic anemias, Fanconi anemia, paroxysmal nocturnal hemoglobinuria, rare cases of acute leukemia, and in normal adults over the age of 70 years. Hypercellularity (cellularity >75%) may be seen in a variety of conditions, including hemolytic anemia, hemorrhage, megaloblastic and sideroblastic anemias, the congenital dyserythropoietic anemias, polycythemia vera and other chronic myeloproliferative neoplasms (MPN), infections, malignant disease, myelodysplastic syndromes (MDS), the leukemias and in normal infants and young children.
Alterations in the frequency and morphology of various types of bone marrow cells
Erythroblasts and neutrophil precursors
Myeloid : erythroid ratio
The myeloid : erythroid ratio (M : E ratio) is often defined as the ratio between the number of cells of the neutrophil granulocyte series (including mature granulocytes) and the number of erythroblasts. The normal range for this ratio in adults has been reported as 2.0–8.3 and 1.1–5.2 in BM smears and 1.5–3.0 in histologic sections.1,2 Some hematologists include eosinophils, basophils and monocytes, and their precursors in the ‘myeloid’ figure but this has only a slight effect on the values for the M : E ratio in normal subjects. The M : E ratio can be used as an index of total erythropoietic activity in patients in whom there is reason to assume that the total number of marrow granulocytes and their precursors is normal (e.g. in patients with normal counts of circulating granulocytes). Conversely, the M : E ratio may be used as an index of total granulocytopoietic activity, provided that the total number of erythroblasts in the body can be assumed to be normal. Some causes of a reduced or increased M : E ratio are listed in Box 5.1.
Morphologic changes in erythroblasts
Most of the erythroblasts in normal BM are uninucleate, show synchrony between nuclear and cytoplasmic maturity and do not display any peculiar cytologic features. In a study of normal BM, only 0.31% of erythroblasts were binucleate (range 0–0.57%), 0.24% (range 0–0.91%) of normal erythroblasts showed basophilic stippling of the cytoplasm, up to 0.7% had vacuolated cytoplasm, 2.38% (range 0.72–4.77%) had intererythroblastic cytoplasmic bridges and 0.22% (range 0–0.55%) possessed markedly irregular or karyorrhectic nuclei.3 Howell–Jolly bodies were found in 0.18% (range 0–0.39%) and asynchrony between nuclear and cytoplasmic maturation was found very infrequently. In various diseases accompanied by a disturbance of erythropoiesis, the frequency of such cytologic features (’dysplastic’ changes) is increased; hence these cytologic ‘aberrations’ are described as dyserythropoietic changes. Morphologic abnormalities that are not encountered in normal BM, such as internuclear chromatin bridges and giant erythroblasts, may also be found in a few diseases (Table 5.1). Dyserythropoiesis occurs in a number of congenital and acquired disorders. The congenital disorders include some thalassemia syndromes, homozygosity for hemoglobin C or E, some unstable hemoglobins, hereditary sideroblastic anemias, thiamine-responsive anemia, homozygosity for pyruvate kinase deficiency, and the congenital dyserythropoietic anemias. The acquired disorders include vitamin B12 and folate deficiency, iron deficiency anemia, alcohol abuse, MDS, acute myeloid leukemia (AML), aplastic anemia, paroxysmal nocturnal hemoglobinuria, acquired immunodeficiency syndrome (AIDS), Plasmodium falciparum and P. vivax malaria, kala azar and liver disease. Dyserythropoietic changes have also been reported after excess ingestion of kelp tablets (containing arsenic) and after BM transplantation.
Table 5.1 Morphologic abnormalities in erythroblasts that may be detected in pathologic states using the light microscope
| Feature | Abnormality | Examples of causative disorder |
|---|---|---|
| Cell size | Large | Megaloblastic anemia Parvovirus B19 infection Congenital dyserythropoietic anemia |
| Small (micronormoblast) | Iron deficiency anemia Thalassaemia Anemia of chronic disease (when severe) Congenital sideroblastic anemia | |
| Nuclei | N : C asynchrony | Megaloblastic anemia Myelodysplastic syndrome and erythroleukemia |
| Irregular shape | Myelodysplastic syndrome Non-neoplastic dysplasia Congenital dyserythropoietic anemia | |
| Nuclear bridges | Myelodysplastic syndrome Congenital dyserythropoietic anemia | |
| Multinuclearity | Myelodysplastic syndrome and erythroleukaemia Congenital dyserythropoietic anemia | |
| Karyorrhexis | Myelodysplastic syndrome Non-neoplastic dysplasia | |
| Cytoplasm | Vacuolation | Alcohol Sideroblastic anemia Chloramphenicol Copper deficiency |
| Cytoplasmic bridging | Myelodysplastic syndrome Congenital dyserythropoietic anemia | |
| Basophilic stippling | Myelodysplastic syndrome Thalassaemia | |
| Sideroblasts | Reduced | Iron deficiency Anemia of chronic disease |
| Increased | Iron overload Megaloblastic anemia Myelodysplastic syndrome | |
| Ring sideroblasts | Congenital sideroblastic anemia Mitochondrial cytopathy (Pearson syndrome) Myelodysplastic syndrome Lead poisoning Drug-induced sideroblastic anaemia |
Increased vacuolation of the cytoplasm of erythropoietic cells has been observed during treatment with chloramphenicol and as an effect of taking excess ethanol. It has also been reported in aplastic anemia associated with glue sniffing, in protein-energy malnutrition, riboflavin and phenylalanine deficiency, AML and hyperosmolar diabetic coma.4 Increased vacuolation of both erythroid cells and granulocyte precursors is observed in Pearson syndrome (a mitochondrial cytopathy) and in copper deficiency.
When there is asynchrony between nuclear and cytoplasmic maturation in a substantial proportion of erythroblasts, erythropoiesis is described as being megaloblastic. A detailed description and the causes of megaloblastic erythropoiesis are given in Chapter 12.
Morphologic changes in the neutrophil series
Morphological abnormalities in the neutrophil series may be of cell size, or shape of nucleus or cytoplasm. There may be abnormalities in the differentiation pathway with left shift or maturation arrest. These changes may be inherited or acquired; the latter may be the result of hematinic deficiencies, infections, drugs, cytokine administration or malignancy. Cytoplasmic abnormalities include the absence of specific granules in the myelocytes and metamyelocytes in acute leukemia and MDS, the reduction of nuclear segmentation in the marrow granulocytes in cases of the inherited and acquired Pelger–Huët anomaly and the formation of giant metamyelocytes and macropolycytes in vitamin B12 or folate deficiency. Giant metamyelocytes may also be found, usually in small numbers, in the absence of evidence of vitamin B12 or folate deficiency, in iron deficiency, infections, malignant disease, falciparum malaria (especially chronic falciparum malaria) and protein-energy malnutrition. They are seen quite often in the BM of patients with AIDS.5 Macropolycytes are also not specific for vitamin B12 or folate deficiency, being found in infections, myeloproliferative neoplasms, drug-induced marrow damage and protein-energy malnutrition. Increased numbers of binucleate cells of the neutrophil series occur in protein-energy malnutrition and to a lesser extent in vitamin B12 or folate deficiency. Macropolycytes and binucleate cells are sometimes seen in MDS. An increased proportion of neutrophils with ring- or doughnut-shaped nuclei may be seen in chronic myelogenous leukemia (CML), AML, MDS, AIDS and falciparum malaria.
Vacuolation of the neutrophil precursors, usually from the promyelocyte/myelocyte stage onwards, may be seen in patients with acute alcoholic intoxication, severe infections, drug-induced marrow damage (e.g. chloramphenicol toxicity), protein-energy malnutrition and certain rare conditions such as the Chédiak–Higashi syndrome, severe congenital neutropenia, hereditary transcobalamin II deficiency,6 neutral lipid storage disease with and without ichthyosis and carnitine deficiency. The term Jordan’s anomaly is sometimes used to designate familial vacuolation of leukocytes; Jordan’s original patient may have had carnitine deficiency.7 Giant metamyelocytes, whatever the condition with which they are associated, may also be vacuolated.
Eosinophil series
An increase of eosinophils and their precursors, sometimes without an associated eosinophilia in the peripheral blood (PB), may be seen in parasitic infections, allergic disorders, certain skin diseases, Hodgkin lymphoma, non-Hodgkin lymphoma, acute lymphoblastic leukemia (ALL), carcinoma, sarcoma and collagen vascular diseases. A BM and PB eosinophilia is also seen in CML, eosinophilic leukemias including those associated with rearrangement of PDGFRA, PDGFRB and FGFR1 (see Chapter 25), other chronic myeloproliferative neoplasms, occasional cases of AML and the idiopathic hypereosinophilic syndrome.
Mast cells
Mast cells may be increased in the marrow in infection and inflammation, renal failure, aplastic anemia, paroxysmal nocturnal hemoglobinuria, lymphoplasmacytic lymphoma including Waldenström macroglobulinemia, chronic lymphocytic leukemia, non-Hodgkin lymphoma, MDS, scleroderma, systemic mastocytosis (in which the mast cells are neoplastic; see Chapter 26), some chronic eosinophilic leukemias (see Chapter 25) and less often in a variety of other conditions. Mast cells are difficult to recognize in sections stained with H&E but are detectable when sections are stained with a Giemsa stain, which stains the granules purple. The granules also stain by the periodic acid-Schiff (PAS) reaction, are α-naphthyl AS-D chloroacetate esterase-positive (positive Leder stain) and stain metachromatically with toluidine blue. They are readily identified with an immunohistochemical stain for mast cell tryptase and may also express mast cell chymotryptase, CD68 and CD117.
Megakaryocytes
Conditions associated with an increased number of megakaryocytes include CML, polycythemia vera, essential thrombocythemia, primary myelofibrosis, infections, chronic alcoholism, reactive changes due to generalized malignant disease, Hodgkin lymphoma, non-Hodgkin lymphoma and hemorrhage. Megakaryocytes are also increased in diseases such as ‘idiopathic’ (autoimmune) thrombocytopenic purpura and thrombotic thrombocytopenic purpura in which thrombocytopenia is primarily caused by a reduced platelet life span (see Chapter 33).
A decreased number of megakaryocytes is seen in acute leukemia, Fanconi anemia and other constitutional aplastic anemias, the syndrome of thrombocytopenia with absent radii, and acquired aplastic anemia. The morphological abnormalities that may affect megakaryocytes in HIV infection are described later in this chapter. Those found in inherited disorders are reviewed in Chapter 32 and those in the myeloproliferative neoplasms in Chapters 22–27.
Lymphocytes
Lymphocytes are more numerous in the BM in children than in adults (Chapters 2 and 3). An artifactual increase in the percentage of lymphocytes occurs if a BM aspirate is much diluted with PB. BM lymphocytes are increased in many reactive and malignant conditions in which there is a PB lymphocytosis (e.g. infectious mononucleosis, chronic lymphocytic leukemia). In addition, BM lymphocytes may be increased in the absence of a PB lymphocytosis in non-Hodgkin lymphoma. The majority of lymphocytes in normal BM are T cells, but BM infiltration is more likely to be seen in B-lymphoproliferative disorders.
A trephine biopsy will show whether a lymphocytic infiltrate is diffuse or focal and whether any focal infiltration is paratrabecular or nodular (with or without follicle formation) (see Chapter 3). In normal BM, lymphocytes are spread diffusely through the marrow but small aggregates or nodules also develop, and rarely they may have germinal centers.8 The incidence of such lymphoid aggregates rises with age, and an increased incidence is seen in pernicious anemia, chronic myeloproliferative neoplasms, hemolytic states, inflammatory reactions and autoimmune conditions such as rheumatoid arthritis and secondary to immunotherapy (e.g. rituximab treatment of lymphoma patients). BM biopsy specimens showing lymphoid aggregates are more likely than other BM specimens to show lipid granulomas and plasmacytosis. A malignant lymphocytic infiltrate may be diffuse or focal and occasionally a nodular pattern is seen (detailed description in Chapters 28 and 29).
Plasma cells
There are less than 1–2% of plasma cells in normal BM. An increased percentage of BM plasma cells may be found in a wide range of pathologic conditions (Box 5.2). In various non-neoplastic conditions, up to 50% of nucleated BM cells may be plasma cells (reactive plasmacytosis). It is sometimes difficult to distinguish between reactive plasmacytosis and multiple myeloma on the basis of the morphologic characteristics of the plasma cells and definitive diagnosis requires immunohistochemistry.9 Some features useful in differential diagnosis are listed in Table 5.2. Russell bodies, Mott cells (plasma cells containing multiple Russell bodies) and apparently intranuclear inclusions resembling Russell bodies (Dutcher–Fahey bodies) may be seen in both reactive conditions and multiple myeloma but apparently intranuclear inclusions (which represent invagination of a cytoplasmic inclusion into the nucleus) are more often seen in myeloma (see Chapter 30). Cells with flaming cytoplasm may be found in both reactive plasmacytosis and myeloma but a substantial proportion of such cells is more likely in myeloma. Examination of the distribution of plasma cells in histologic sections of BM is of considerable value in elucidating the cause of plasmacytosis since homogeneous nodules of these cells, with little supporting stroma, are found in myeloma but not in reactive plasmacytosis (Table 5.2). Myeloma cells may show hemophagocytosis. Hemosiderin-containing granules are sometimes found in the cytoplasm of plasma cells in alcoholics and in copper deficiency, porphyria cutanea tarda, megaloblastic anemia, refractory normoblastic anemia and iron overload.
Box 5.2 Causes of plasmacytosis in the bone marrow
Table 5.2 Characteristics of bone marrow plasma cells in reactive plasmacytosis and multiple myeloma
| Reactive plasmacytosis | Multiple myeloma | |
|---|---|---|
| Number | Up to 10–20%, rarely up to 50% | Usually 30–90% |
| Cytology | Most cells are mature and look like normal plasma cells Majority of cells are mononucleate; four nuclei per cell rare Nucleoli only in occasional cells Nucleocytoplasmic asynchrony usually not a prominent feature | More cells show features of immaturity; cells are either pleomorphic or monomorphic; occasionally lymphoid Multinuclearity common Nucleoli common Nucleocytoplasmic asynchrony common |
| Distribution | Interstitial infiltrate, especially perivascular; some cells may be aggregated around macrophages Small clusters of plasma cells may be present but large homogeneous nodules are absent Broad band-like infiltrates very rare | Commonly near endosteal surface as well as perivascular Large homogeneous nodules of plasma cells with little intervening hemopoietic tissue common Broad band-like infiltrates common |
| Immunocytochemistry | κ : λ ratio about 2 : 1 CD19 positive CD56 negative | Monotypic κ- or λ-chains CD19 negative CD56 positive (90% cases) |
Macrophages (histiocytes)
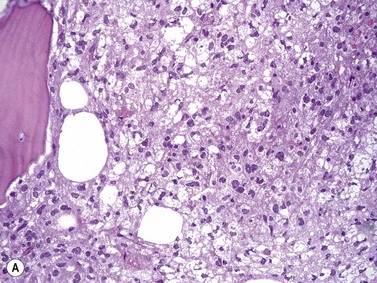
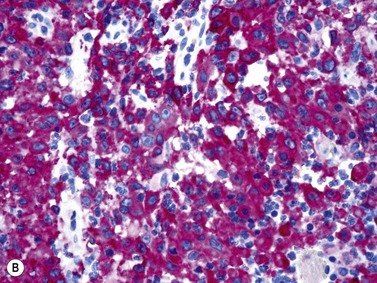
An increase of BM macrophages is common in a wide variety of hematologic and non-hematologic conditions. These include various infective and inflammatory disorders, conditions associated with increased blood cell destruction or increased ineffective hemopoiesis and post-granulocyte-macrophage colony-stimulating factor (GM-CSF) therapy. The macrophages range from immature cells showing little phagocytic activity to mature cells containing phagocytosed material or having foamy cytoplasm. In most instances the increase in macrophages is reactive. However, in the rare neoplastic condition designated malignant histiocytosis the increase results from the proliferation of cells of a neoplastic clone.10 The histiocytic disorders are defined by their constitutive cell type11 (Box 5.3). The two morphological features that distinguish malignant histiocytosis from reactive macrophage hyperplasia are: 1) pleomorphism of macrophages, with immature and atypical features such as a prominent nucleolus, distinct and thick nuclear membrane, irregular nuclear chromatin and multinuclearity; and 2) the presence among the macrophages and large multinucleate cells of many monoblasts and promonocytes12 (Fig. 5.1). The number of malignant cells in the BM varies from 5 to 90%12 and the infiltration of the marrow may be focal or diffuse (Fig. 5.2). The demonstration of positivity for CD163 (to differentiate from other mesenchymal tumors that may express CD68 shared by monocytes/macrophages) and a clonal cytogenetic abnormality in BM cells provides strong supporting evidence. Cytochemical reactions of the malignant cells are similar to those of monocytes and macrophages; they are positive for nonspecific esterase, acid phosphatase and lysozyme. Immunohistochemical analysis distinguishes tumors derived from Langerhans cells (Langerhans cell histicytosis and sarcoma), follicular dendritic cell sarcoma and interdigitating cell sarcoma from true histocytic sarcoma and malignant histiocytosis.13 Primary bone marrow involvement in these tumors is relatively rare. The blood count may show anemia, leukopenia, thrombocytopenia and eosinophilia and the PB film may contain macrophages and small numbers of monoblasts. A minor degree of hemophagocytosis is common. It is now apparent that the majority of patients initially described as having malignant histiocytosis actually had a reactive hemophagocytic syndrome, which could have been associated with malignant lymphoma and/or Epstein–Barr virus (EBV) infection.14–16
Box 5.3 Classification of benign histiocytic disorders
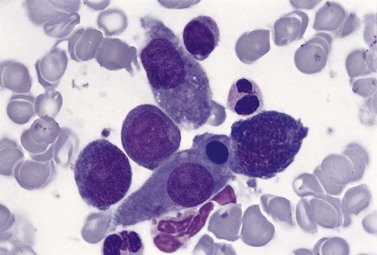
Hemophagocytic syndromes
The hemophagocytic syndrome, also called hemophagocytic lymphohistiocytosis (HLH), is a collection of non-malignant but frequently life-threatening disorders, associated with an ever growing list of genetic and acquired causes17,18 (Box 5.4). In HLH, there is a deregulation of T-lymphocytes and excessive production of cytokines leading to macrophage hyperplasia, enhanced macrophage activity and increased phagocytosis by macrophages of red cells, granulocytes, platelets and hemopoietic cells. The clinico-pathologic features of the syndrome include fever, hepatosplenomegaly, lymphadenopathy, skin rash, neurologic abnormalities, cytopenias, hypertriglyceridemia, high serum ferritin level and coagulopathy. In the ‘primary’ hemophagocytic syndrome (familial hemophagocytic lymphohistiocytosis), there is an inherited immune defect. Many cases of acquired hemophagocytic syndrome have an underlying predisposing condition leading to immunosuppression such as HIV infection, renal transplantation, malignant disease and autoimmune disease. The most frequent cause of a virus-associated hemophagocytic syndrome is EBV. The many other causes of infection-associated hemophagocytic syndrome include tuberculosis and P. falciparum malaria (Fig. 5.3). Reactive macrophages containing phagocytosed blood or BM cells can be distinguished on the basis of cytologic and cytochemical characteristics from other malignant cells showing hemophagocytic activity (Box 5.4).



