CHAPTER 33 Acquired disorders affecting megakaryocytes and platelets
Structure and function of megakaryocytes and platelets
Human platelets
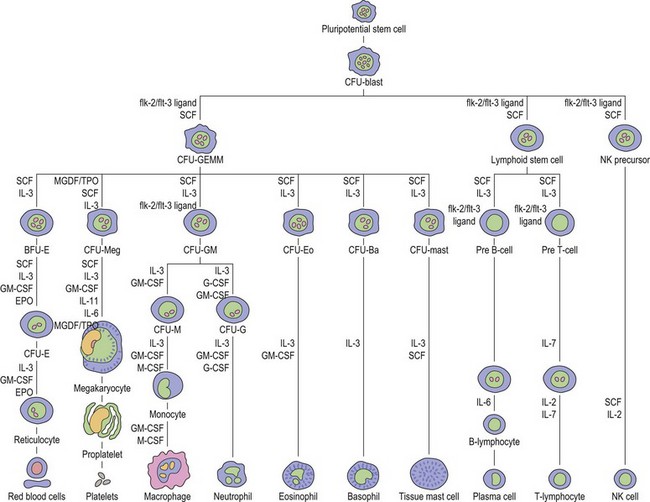
Thrombopoiesis, the generation of platelets from megakaryocytes in the bone marrow, is complex and incompletely understood. Megakaryocytes are large end-stage cells from which platelets bud. The earliest recognized committed progenitor is the burst-forming unit (BFU)-Meg.1 Fig. 33.1 shows megakaryocyte development from stem cell stage through to platelet production. BFU-Megs develop into colony-forming unit (CFU)-Megs in the presence of growth factors thrombopoietin (TPO), interleukin-3 (IL-3) and IL-11. Megakaryocyte nuclei are large polyploid structures with chromosome contents between diploid (2N) to 64N. Such polyploid status is achieved through a process termed nuclear endoduplication: that is, successive doubling of chromosome content in the absence of cell division. Platelets are produced from megakaryocytes that are 8N or greater.2 A single megakaryocyte can generate around 3000 platelets of which 20–30% are pooled in the spleen. In health the peripheral blood platelet count is 150–400 × 109/l but this fluctuates, for example, following heavy exercise, ‘stress’, and around the menstrual cycle. This transient rise in platelet count may be caused by mobilization of platelets pooled in the spleen. There are also racial differences in the ‘normal’ platelet count and some Mediterranean populations have platelet counts as low as 80 × 109/l in health. Platelets are produced at a rate of 35 000–44 000 per microliter per day3 and have a lifespan of 9–10 days.
Platelet structure and function
The integrin family of proteins
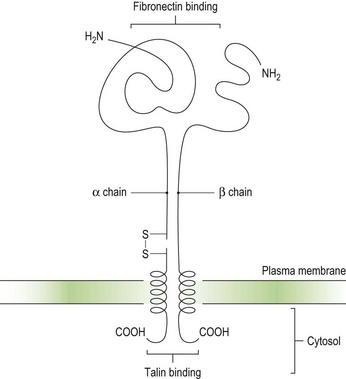
Integrins are key platelet membrane proteins and have been characterized on a large number of leukocytes and many other cells. For example, the fibronectin receptor on mammalian fibroblasts is one of the best characterized matrix receptor proteins4 (Fig. 33.2). This receptor, in common with all other integrins, is a heterodimer consisting of a non-covalently associated complex of two distinct high-molecular-weight polypeptides, α and β. The receptor functions as a transmembrane linker which mediates the interaction between the intracellular actin cytoskeleton and fibronectin in the extracellular matrix. Like all integrins, the fibronectin receptor recognizes so-called RGD (Arg-Gly-Asp) sequences in matrix components. In platelets, integrins recognize and bind a variety of proteins in order to form a hemostatic plug through a complex mechanism of platelet adhesion, shape change and activation of the clotting pathway.
Platelet integrins and related proteins
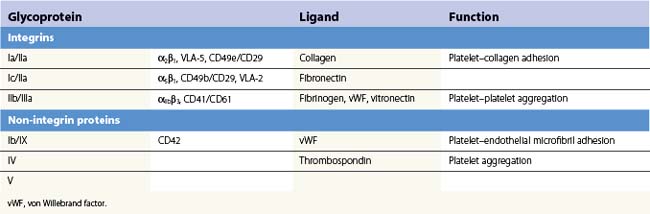
Platelets contain five integrin α subunits and two β subunits producing: αIIbβ3, ανβ3, α2β1, α5β1 and α6β1.5,6,7 These are shown in Fig. 33.3. Further detail is provided by Table 33.1. These proteins are essential for normal platelet function, and are often the target of immunological attack in disorders such as idiopathic thrombocytopenic purpura (ITP).
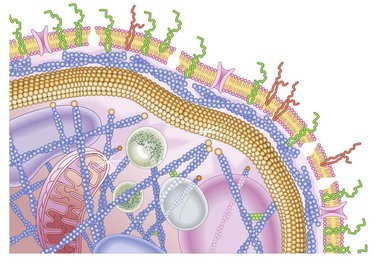
Fig. 33.3 Cartoon of platelet membrane, showing platelet glycoproteins.
(Courtesy of Peg Berrity, Science Photo Library.)
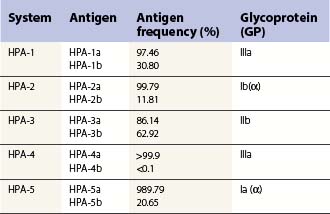
Platelet alloantigens
These can be platelet-specific or shared with other cells. Important shared antigens include HLA class I and ABH (blood group A and B) antigens. Platelet-specific antigens fall into five well-defined human platelet antigen (HPA) groups (Table 33.2; see also Chapter 37): HPA-1, HPA-2, HPA-3, HPA-4 and HPA-5, each of which has an α and β allele. Each platelet allotype represents a single amino acid substitution in the platelet glycoprotein molecule. Because some platelet glycoproteins carry epitopes that play a major role in platelet function, platelet alloantibodies may not only cause thrombocytopenia but also affect primary hemostasis.
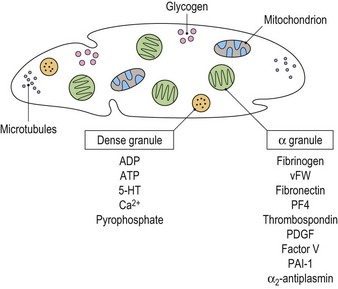
Cytoplasmic platelet constituents
Platelets contain two principal types of granule: dense bodies and α granules (Fig. 33.4). Dense bodies contain ADP, ATP, 5-HT, calcium and pyrophosphate. The α granules contain more than 300 releasable proteins including adhesion molecules, chemokines, cytokines, coagulation factors, fibrinolytic regulators, growth factors and pro- and anti-angiogenic factors such as PF4, β-thrombospondin, PDGF, vWF, fibrinogen, factor V and fibronectin. The contents of these granules are integral components of the platelet’s biological activities.
Biological function of platelets
The primary role of the platelet is the prevention of blood loss from damaged tissues and vessels, i.e. primary hemostasis. This is achieved through platelet activation, adhesion, shape change and aggregation. Platelets may also play a role in the maintenance of vascular integrity by the constitutive release of cytokines and growth factors from their granules that bind to endothelial cell surface receptors resulting in intracellular signaling that stabilizes the molecular complexes that form the junctions between adjacent endothelial cells.8
Platelet adhesion (Fig. 33.5A)
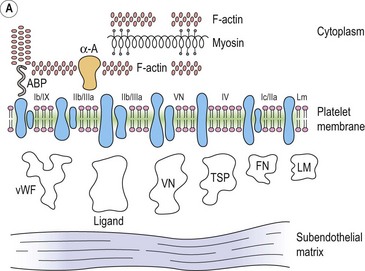
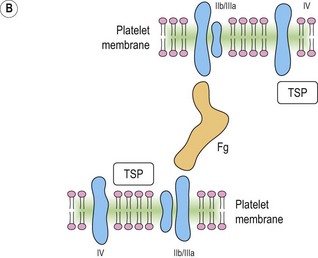
Platelet shape change
After adhesion to the subendothelium, platelets undergo a major shape change, from a discoid shape to one which is irregular, with projections (Fig. 33.6). This process is initially reversible but ultimately becomes irreversible.
Quantitative platelet abnormalities: thrombocytopenia
Thrombocytopenia, a reduction in platelet count, may be caused by:
Pseudothrombocytopenia
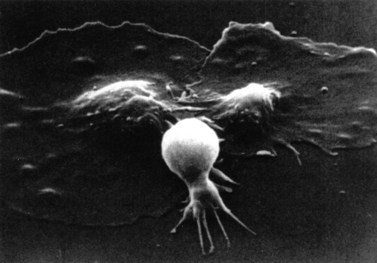
This describes patients in whom the peripheral blood platelet count is found to be spuriously low, and is caused by a variety of mechanisms. Platelet clumping caused by the anticoagulant ethylenediaminetetraacetic acid (EDTA) is the commonest cause, with an incidence of around 0.1%. EDTA-induced pseudothrombocytopenia is easily excluded by examination of a blood film which will confirm the presence of numerous large platelet clumps.9,10 Other causes of pseudothrombocytopenia include the presence of giant platelets, which are not counted as platelets by modern automated counters, and platelet satellitism (platelets attach themselves to monocytes or granulocytes) (Fig. 33.7).
Pooling of platelets in the spleen
Mechanism involved
In health, the spleen may pool up to one third of the total platelet mass, and in disease states this may rise to 90%.11 Although the peripheral blood platelet count may only be a fraction of the normal range, the patient generally has an overall normal platelet mass since production is entirely normal but the low peripheral counts simply reflect a larger than normal mass of platelets pooled in the spleen.
Thrombocytopenia due to failure of platelet production
Acquired amegakaryocytic thrombocytopenia
This describes severe thrombocytopenia caused by a selective reduction in megakaryocytes in an otherwise normal bone marrow, and is the result of damage to the megakaryocyte stem cell. The disorder is analogous to acquired pure red-cell aplasia (PRCA). Acquired amegakaryocytic thrombocytopenia may be caused by drugs, toxins and connective tissue disorders.12–14
Bone marrow failure syndromes
The bone marrow failure (BMF) disorders are those in which there is a failure of bone marrow precursor (stem) cells in contrast to those disorders such as myelodysplastic syndromes in which normal or increased numbers of abnormal cells are produced, or those in which the survival of the cells is reduced. The BMF syndromes are a diverse groups of disorders with a common endpoint in which there is loss of hematopoietic stem cells. BMF can be acquired or inherited (see Table 33.3).
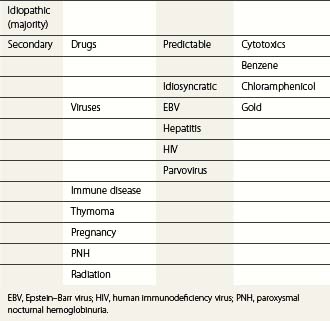
Thrombocytopenia due to increased platelet destruction
Causes may be immunologic or non-immunologic.
Non-immunologic causes of thrombocytopenia
Disseminated intravascular coagulation (DIC)
DIC is characterized by excessive activation of the coagulation cascade (see also Chapter 35). In most cases DIC is an acute event, but chronic DIC is well described, although clinically less important. The main problem faced in patients with DIC is bleeding, which may be mild but is often severe with generalized oozing from venepuncture sites, central lines and other indwelling cannulae, gastrointestinal and genitourinary tracts. Microthrombi are found in 5–10% of cases, often affecting digits, with resulting peripheral gangrene.
Pathogenesis
DIC is triggered by the release or exposure of tissue thromboplastins which contain a high concentration of phospholipids following trauma, surgery, mismatched blood transfusion and a variety of other triggers (Table 33.4). In addition to systemic activation of coagulation (leading to fibrin clot formation, organ failure and consumption of platelets and coagulation factors that may cause bleeding), there is dysregulation of natural anticoagulant systems and fibrinolysis.
Table 33.4 Triggers for disseminated intravascular coagulation (DIC)
| Trauma | Including surgical |
| Dissemination of cancer cells | Malignancy, following administration of chemotherapy |
| Massive hemolysis | Post mismatched blood transfusion |
| Venoms | e.g. snake venoms |
| Endothelial injury | Gram-negative sepsis |
| Infections | |
| Burns | |
| Septicemia |
Thrombotic thrombocytopenic purpura and hemolytic uremic syndrome
Thrombotic microangiopathy refers to a state that is characterized pathologically by occlusive microvascular thrombosis and clinically by profound thrombocytopenia, microangiopathic hemolytic anemia and variable signs and symptoms of organ ischemia.16 The term primarily refers to two discrete but overlapping syndromes, thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS).
TTP is a disseminated microangiopathy, first described by Moschowitz in 1924.17 It is uncommon, occurring primarily in adults and with an annual incidence in the US of 4–11 cases per million people. HUS has clinical and laboratory features that often overlap with those of TTP. However, HUS is more common in children, the renal abnormalities are more marked than in TTP (Table 33.5; see also Chapter 10) and the underlying pathogenetic mechanisms of the two conditions differ.
Table 33.5 Classification of thrombocytopenic purpura and hemolytic uremic syndrome.
| Idiopathic TTP/HUS | Classic adult TTP and childhood non-verotoxin-associated HUS-TTP | |
| Secondary TTP-HUS | Pregnancy-related | TTP, postpartum HUS |
| Verotoxin-induced | Escherichia coli and Shigella dysenteriae I | |
| Childhood HUS | ||
| Epidemic adult TTP-HUS | ||
| Malignant disease | Especially metastatic carcinomas | |
| Drug-induced | Chemotherapy agents, e.g. mitomycin C, cisplatin, and other drugs | |
| Immunosuppressive agents, e.g. cyclosporin, quinine, ticlopidine | ||
| Post-marrow/stem cell transplantation | Especially in conjunction with total body irradiation or high-dose (intensive) chemotherapy |
HUS, hemolytic uremic syndrome; TTP, thrombocytopenic purpura.
(Modified from George JN, El-Harake M 1995. Thrombocytopenia due to enhanced platelet destruction by non-immunologic mechanisms. In: Beutler E, Lichtman MA, Coller BS, Kipps TJ (eds), Williams Hematology, 5th edn. McGraw-Hill, New York, 1290–1315)
Clinical features of TTP
TTP is characterized by the pentad:
TTP typically has a sudden onset, with fever and neurologic symptoms including paralysis, coma, fits and psychiatric disturbance. There is usually purpura and the clinical picture is one of a fluctuating course. The causes of TTP can be subclassified into: 1) congenital; 2) idiopathic; and 3) non-idiopathic.16
Laboratory investigations
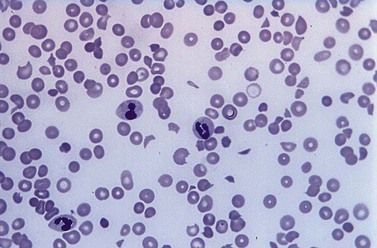
A full blood count and blood film will show anemia (hemoglobin ~ 8–9 g/dl) with polychromasia and other evidence of hemolysis. The film will usually show red-cell fragments and thrombocytopenia (Fig. 33.8). There is usually evidence of hemoglobinemia reflecting the presence of intravascular hemolysis. Lactate dehydrogenase will be elevated. Clotting tests are generally normal although occasionally there may be features of DIC. The direct Coombs test is negative. Renal failure is uncommon but elevated serum creatinine is seen in about a third of cases.
Pathogenesis
Failure to cleave large von Willebrand factor (vWF) multimers is thought to be crucial in the pathogenesis of TTP. Von Willebrand factor is synthesized in vascular endothelial cells and megakaryocytes and assembles into multimers linked by disulfide bonds which are stored in the Weibel–Palade bodies of endothelial cells and α-granules of platelets. A small proportion of these multimers is constitutively secreted by endothelial cells into the circulation, stabilizing factor VIII and mediating both platelet adhesion to sites of vascular injury and platelet aggregation in conditions of high shear. Endothelial cell stimulation causes unusually large vWF multimers to be secreted and these attach to the endothelial cell surface.16
Management
Plasma exchange with cryosupernatant or FFP is now regarded as the treatment of choice for TTP and should be instituted as soon as possible after diagnosis and continued on a daily basis with replacement of 1.0 to 1.5 times the predicted plasma volume of the patient. Plasma exchange should continue until there is normalization of the neurological state, the platelet count has been >150 × 109/l for at least 2 days, the LDH is normal and the hemoglobin rising.18 The disease is highly variable in its course and it is difficult to predict the clinical outcome at the outset. Patients require intensive care nursing, and may require ventilation. Hemodialysis may be required along with plasma exchange. A short course of high dose corticosteroids has also been recommended along with aspirin when the platelet count has recovered to 50 × 109/l.18 Platelet transfusions should be avoided except in cases of severe hemorrhage. Remission is achieved when the platelet count remains normal for 30 days after discontinuation of plasma exchange.19 Patients with severe deficiency of ADAMTS 13 appear to have an increased risk of relapse, most within a year. Rituximab has been used to try to reduce the risk of relapse.20,21
Hemolytic uremic syndrome
The disorder was first described in 1955 by Gasser,22 who reported on five patients (all infants) with acute renal failure, who died of renal cortical necrosis. HUS is classified into D+HUS and D-HUS.16 D+HUS accounts for >90% of cases and is caused by enteric infection with Shiga toxin-producing bacteria, most commonly Escherichia coli O157:H7 or occasionally Shigella dysenteriae I.22 It can occur at all ages but is seen typically in young children.22–24 HUS usually develops 4–6 days after the onset of diarrhea. There is a seasonal incidence, with the highest number of new cases reported in the summer months.25,26
D-HUS is uncommon and clinically heterogeneous. These individuals do not have an antecedent enteric infection with a Shiga toxin-producing organism and have a relatively poor prognosis with a mortality rate of about 25%.16
The histopathological features of HUS are different from those of TTP. The kidneys are preferentially involved and the thrombi are composed primarily of fibrin with few platelets and little vWF.16 Endothelial damage is pronounced in childhood D+HUS. Marked destruction of the renal cortex may occur and glomerular thrombosis is characteristic with an appearance suggesting capillary congestion rather than ischemia.16 D-HUS has been little studied but the renal lesions appear to differ with more pronounced mesangial involvement and less glomerular thrombosis than in D+HUS.16
Pathogenesis. Most D+HUS results from E. coli O157 infections caused by food-borne outbreaks from cattle. Unwashed fruit is another source of some outbreaks. Subunits of Shiga toxin produced by the bacteria bind to microvascular endothelial cells particularly in the kidney and on monocytes and platelets. The toxin is subsequently internalized and inhibits protein synthesis leading to cell death. Cytokine release is stimulated further increasing Shiga toxin binding. Resulting endothelial cell damage has prothrombotic consequences that lead to the histopathological changes described above.16
D-HUS in contrast is associated with complement dysregulation and mutations in one of the complement regulatory proteins (factor H, factor I, membrane cofactor protein (MCP) and factor B) have been described in about 50% of these patients.16 In some patients an autoantibody against factor H has been recognized and in about 5% of cases mutations that impair thrombomodulin function have been identified.27
Laboratory findings. There is evidence of acute renal failure and features of microangiopathic hemolysis.28 The blood count will generally show an elevated WBC, most usually neutrophilia; the prognosis is worst for patients with total neutrophil counts exceeding 20 × 109/l.25
Outlook. Unlike adult TTP, childhood D+HUS is rarely fatal and the mortality is around 3–5%. The outlook is less favorable in D-HUS. In cases with end-stage renal failure that have undergone renal transplantation, the outcome appears to correlate with the cause of the complement dysregulation. Recurrence of HUS has not been seen in transplanted kidneys in patients with MCP mutations whereas the condition did recur following renal transplantation in patients with factor H or factor I mutations, perhaps because these latter two factors are made in the liver. Combined liver and kidney transplants may be successful in some of these cases.16
Pre-eclampsia and HELLP syndrome
In addition to TTP and HUS there are a number of causes of thrombotic microangiopathy that can occur during pregnancy, in particular pre-eclampsia/eclampsia and the HELLP syndrome. Differentiation between these different conditions can be problematic. Pre-eclampsia occurs after 20 weeks of pregnancy and is associated with hypertension, edema, sodium retention, proteinuria and DIC. It may progress to eclampsia which is characterized by convulsions.29 Previously, pre-eclampsia/eclampsia and HELLP have been considered distinct disorders but it seems more likely that they represent a spectrum of a pathologic process.
HELLP syndrome
This disorder, characterized by hemolysis, elevated liver enzymes and low platelet count, occurs in 0.5–0.9% of all pregnancies,30,31 and is associated with a mortality rate of 1–4%.32 It occurs in up to 10% of cases of severe pre-eclampsia. Perinatal mortality is high and may reach 10–20%.32
Clinical features. HELLP is associated with generalized weakness, nausea and vomiting, right upper quadrant pain, headache and visual upset.33 In some cases HELLP may occur following delivery.36
Pathophysiology. There may be abnormalities of the placental vessels with resulting placental ischemia. This is associated with the systemic release of a thromboxanes, angiotensin, tumor necrosis factor-α (TNF-α) and other procoagulant proteins.31,33 DIC may result leading to thrombi which threaten major end-organs including placenta, renal, hepatic and central nervous systems. The thrombi lead to endothelial damage and there is microangiopathic hemolytic anemia (MAHA) and failure of the organs affected by the thrombotic process. Liver failure and occasionally liver rupture may result.34
Management. Prompt delivery, and control of the hypertension are required, in addition to correcting the factor deficiencies caused by the DIC. Corticosteroids have been used as well as plasma exchange and plasmapheresis.35 Maternal deaths, where these occur, are usually due to uncontrolled DIC. Cases in which thrombotic microangiopathy persists post-partum should be considered for plasma exchange.18
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


