Overview
Abnormalities in laboratory tests are frequently the first or only sign of liver disease, and the pattern of abnormality is often suggestive of the underlying disease process. Gamma-glutamyltransferase (GGT) is particularly sensitive for liver disease. If the level of GGT is normal, there is only a 1–2% chance of liver disease. The levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) are also very useful for the diagnosis of liver disease. An AST > 3000 U/L suggests a severe hypotensive episode causing centrilobular necrosis, a toxic injury such as acetaminophen overdose, or acute viral hepatitis. On the other hand, chronic diseases of the liver such as alcoholic liver disease and chronic viral hepatitis are typically associated with smaller elevations of transaminases, in the 100–300 U/L range. Elevated ALT and AST with an AST/ALT ratio > 2:1 is classically associated with alcoholic hepatitis. Elevated alkaline phosphatase (ALP) can be seen in both liver and bone disease, whereas a concomitant elevation of ALP and GGT is consistent with cholestatic liver disease.
While the above enzymes (GGT, AST, ALT and ALP) indicate damage to the hepatocytes, prothrombin time (PT) and serum albumin are more reflective of the functional status of the liver, since both albumin and clotting factors are produced by hepatocytes. Factor VII has a serum-half life of about 4 hours, making the PT a good assessment of an acute change in liver function, whereas albumin is more accurate at assessing a chronic change in liver function. Also, assessment of gamma globulins is useful for determining an acute versus chronic pathologic liver process. In acute processes, the gamma globulin level is normal, and in chronic processes, it is elevated (> 3 g/dL).
When assessing liver function tests, four general patterns are apparent: (1) acute hepatitis pattern, which has elevated transaminase levels and variable increases in other enzymes; (2) cirrhosis pattern, which has decreased albumin, elevated gamma globulins (with β–γ bridging on serum electrophoresis) and elevated PT; (3) chronic hepatitis pattern, which has a combination of changes seen in acute hepatitis and cirrhosis patterns; and (4) obstructive liver disease pattern, also called cholestasis, which has an elevated ALP and bilirubin (Table 15-1).
Patterns of Liver Disease | Laboratory Studies |
|---|---|
Acute hepatitis pattern | ↑ AST, ALT |
Cirrhosis pattern | ↓ albumin, ↑ gamma globulins |
Chronic liver disease pattern | ↑ AST, ALT ↓ albumin, ↑ gamma globulins |
Obstructive liver disease pattern | ↑ ALP, bilirubin |
Ascites is often associated with cirrhosis; however, both transudative ascites (seen in patients with cirrhosis, alcoholic hepatitis, or congestive heart failure) and exudative ascites (seen in patients with peritoneal carcinomatosis or tuberculosis) may occur. Differentiating between the two types requires a determination of gradient (i.e., difference between serum albumin and ascitic fluid albumin). Transudative ascites has a high gradient (> 1.1 g/dL), and exudative ascites has a low gradient (< 1.1 g/dL).
This chapter discusses liver pathology, including jaundice and cholestasis, hepatic failure, cirrhosis, vascular and circulatory disorders, alcoholic liver disease, metabolic diseases, obstructive biliary tract disorders, and hepatic tumors; gallbladder pathology, including gallstones and acute and chronic cholecystitis; and pancreatic pathology, including pancreatitis and pancreatic adenocarcinoma.
General Responses of the Liver to Injury
Overview: Although many different toxins, infections, and other conditions affect the liver, the liver only has five general responses to injury: inflammation, cellular accumulations, cell death, fibrosis, and regeneration. Cirrhosis results from a combination of fibrosis and regeneration after cell death. Of the five general responses of the liver, only cellular accumulations and cell death will be discussed here.
- Macrovesicular steatosis: One large fat vacuole in hepatocytes; associated with alcohol use, obesity, and diabetes mellitus.
- Microvesicular steatosis: Many small vacuoles in hepatocytes; associated with acute fatty liver of pregnancy, Reye syndrome (occurs in children with viral illness when given aspirin), alcohol, and certain drugs (e.g., tetracycline).
- Spotty necrosis: Focal areas of necrosis in the lobular parenchyma.
- Interface hepatitis: Necrosis at the edge of the limiting plate.
- Bridging necrosis: Necrosis spanning between the portal tracts and from the portal tracts to the centrilobular hepatocytes.
- Submassive necrosis: Necrosis of entire lobules.
- Massive necrosis: Necrosis of almost the entire liver.
Jaundice and Cholestasis
Overview: Jaundice is an accumulation of unconjugated or conjugated bilirubin in skin that produces a golden yellow color. Jaundice is evident in skin when the serum bilirubin level is 2.5–3 mg/dL or higher. Yellow discoloration of the sclerae is referred to as icterus (Figure 15-1). Cholestasis is an accumulation of bile. Bile contains much more than just bilirubin, including bile salts and cholesterol.

- Excessive production of bilirubin results in unconjugated hyperbilirubinemia (causes: hemolytic anemias and ineffective erythropoiesis, such as in megaloblastic anemia).
- Reduced hepatic uptake results in unconjugated hyperbilirubinemia (causes: rifampin, which competes for bilirubin uptake; Gilbert syndrome).
- Impaired conjugation results in unconjugated hyperbilirubinemia (causes: physiologic jaundice of the newborn, Crigler-Najjar syndrome, diffuse hepatocellular damage).
- Decreased hepatocellular excretion causes conjugated hyperbilirubinemia (causes: Dubin-Johnson syndrome, diffuse hepatocellular damage).
- Impaired bile flow (from intrahepatic or extrahepatic obstruction) results in conjugated hyperbilirubinemia.
- Causes of intrahepatic obstruction: primary biliary cirrhosis, primary sclerosing cholangitis.
- Causes of extrahepatic obstruction: gallstones, carcinoma of the pancreas, extrahepatic biliary atresia or biliary strictures, and extrahepatic primary sclerosing cholangitis.
- Causes of intrahepatic obstruction: primary biliary cirrhosis, primary sclerosing cholangitis.
Mechanism of Jaundice and Cholestasis | Form of Hyperbilirubinemia | Causes |
|---|---|---|
Increased bilirubin production | Unconjugated | Hemolytic anemia, ineffective hematopoiesis |
Decreased hepatic uptake | Unconjugated | Gilbert syndrome, rifampin use |
Impaired conjugation | Unconjugated | Physiologic jaundice of newborn, Crigler-Najjar syndrome, diffuse hepatocellular disease |
Decreased hepatic excretion | Conjugated | Dubin-Johnson syndrome, diffuse hepatocellular disease |
Impaired intrahepatic bile flow | Conjugated | Primary biliary cirrhosis, primary sclerosing cholangitis |
Impaired extrahepatic bile flow | Conjugated | Gallstone, pancreatic carcinoma |
Overview: Although there are many causes of jaundice and cholestasis, the five conditions that will be discussed in this section are hereditary conditions or conditions associated with the neonate. These conditions are physiologic jaundice of the newborn, Gilbert syndrome, Crigler-Najjar syndrome, Dubin-Johnson syndrome, and neonatal cholestasis.
Basic description: The enzymes that conjugate bilirubin do not mature for a few weeks after birth; therefore, neonates are prone to development of unconjugated hyperbilirubinemia.
Epidemiology: Common (> 5% of the population); male predominance.
Mechanism: Mutations of the UGT gene lead to decreased levels of uridine diphosphate-glucuronyl transferase (at 30% of normal level), which causes decreased ability to conjugate bilirubin with glucuronic acid.
Clinical presentation of Gilbert syndrome: Patients become jaundiced when stressed (e.g., illness, exercise).
Inheritance pattern: Both types I and II are autosomal recessive.
Mechanism: Type I has a complete lack of enzyme needed for the conjugation of glucuronic acid to bilirubin; type II has partial lack of the enzyme.
Mechanism: Absence of canalicular protein multidrug-resistant protein 2 (MRP2), which transports bilirubinglucuronides.
Gross morphology: Black liver.
Causes: Bile duct obstruction, infections (e.g., caused by cytomegalovirus, sepsis), toxins, metabolic (e.g., α1-antitrypsin deficiency), and idiopathic (50% of cases).
Microscopic morphology of neonatal cholestasis: Giant cells, cholestasis, mononuclear cells in portal tracts, and extramedullary hematopoiesis.
Gross morphology of jaundice and cholestasis: Scleral icterus; skin xanthomas due to impaired excretion of cholesterol.
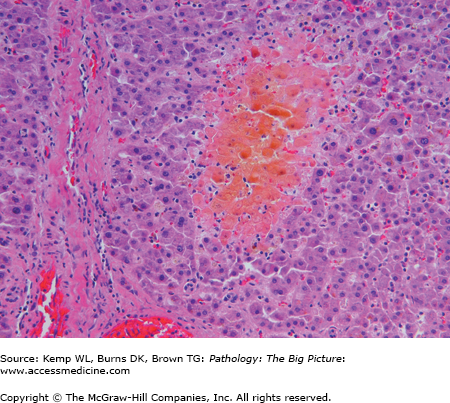
Microscopic morphology of jaundice and cholestasis (Figures 15-2 and 15-3)
- Bile plugs within canaliculi (seen in cholestasis).
- Foamy degeneration of hepatocytes (due to intracellular accumulation of bile).
- If cholestasis is due to intra- or extrahepatic bile duct obstruction:
- Distension of upstream bile ducts; proliferation and edema of bile ducts.
- Bile lakes (i.e., focal destruction of parenchyma with accumulation of bile).
- Cirrhosis due to portal tract fibrosis.
- Distension of upstream bile ducts; proliferation and edema of bile ducts.

Clinical presentation of jaundice and cholestasis
- Symptoms: Patients with cholestasis have pruritus as a result of deposition of bile salts in tissue, and may have clay-colored stools resulting from failure of bile to reach the intestines.
- Signs: Elevated bilirubin (unconjugated or conjugated, depending upon the cause of jaundice); also increased concentration of ALP in cholestasis. ALP is produced by bile duct epithelium and canalicular hepatocytes and is released by detergent action of bile. ALP is increased to three to four times the normal level, and the AST and ALT are increased more than five to ten times the normal levels.
Laboratory studies to differentiate causes of jaundice and cholestasis
- Hemolytic causes: No urine bilirubin, elevated urine urobilinogen, and elevated unconjugated bilirubin.
- Hepatocellular cause: Increased urine bilirubin, normal urine urobilinogen, and elevated unconjugated and conjugated bilirubin.
- Obstructive causes: Increased urine bilirubin, decreased urine urobilinogen, and elevated conjugated bilirubin.
Diagnosis of cause of jaundice and cholestasis: Biopsy of liver; radiographic imaging to determine location and source of obstruction.
Hepatic Failure
Overview: For liver failure to occur, there must be a loss of 80–90% of hepatic parenchyma. Hepatic failure occurs in one of three situations: massive hepatic necrosis, chronic liver disease, or widespread but nonfatal injury to the hepatocytes. Hepatic failure as a result of chronic liver disease is most often due to the end stage of cirrhosis. Widespread damage to hepatocytes, which maintain viability, has several causes, including acute fatty liver of pregnancy. Massive hepatic necrosis as a cause of hepatic failure will be discussed below.
Basic description: Massive hepatic necrosis is often the result of fulminant hepatitis or toxic injury. The definition of fulminant hepatic failure is the onset of encephalopathy within 8 weeks of the onset of jaundice in a patient with hepatic injury and no history of prior liver disease.
Causes of massive hepatic necrosis
- Acetaminophen toxicity (35–40% of cases).
- Other toxins: Halothane, isoniazid.
- Acute hepatitis A (4% of cases).
- Acute hepatitis B (8% of cases).
- Others causes: Acute fatty liver of pregnancy, Wilson disease, hepatic ischemia, Amanita mushroom poisoning.
- Specific cause: Reye syndrome (clinical presentation in children: Reye syndrome presents as vomiting a few days after treatment of a viral infection with aspirin; patients progress to seizures, cloudy sensorium, and coma).
Overview: Include hepatic encephalopathy, hepatorenal syndrome, jaundice, hyperammonemia, coagulopathy, hypoglycemia, and infections. Several of these complications are simply due to loss of normal liver functions. Coagulopathy is a result of deficiency of vitamin K–dependent factors II, VII, IX, and X. Hypoglycemia is a result of impaired gluconeogenesis. Infections are one of the leading causes of death in patients with fulminant hepatic failure. Hepatic encephalopathy and hepatorenal syndrome are discussed below.
Basic description: Complex neuropsychiatric syndrome that complicates advanced liver disease.
Forms: acute and chronic
- Acute hepatic encephalopathy: Occurs in the setting of fulminant hepatic failure. Cerebral edema is more prominent in acute hepatic encephalopathy than in the chronic form.
- Chronic hepatic encephalopathy: Occurs with chronic liver disease; is reversible.
Pathogenesis of hepatic encephalopathy: Inadequate removal of nitrogenous compounds or other toxins that are ingested or formed in the gastrointestinal tract. Common precipitants include gastrointestinal bleeding and gastrointestinal protein loading, portosystemic shunting, and infections.
Microscopic morphology: Characterized by Alzheimer type II astrocytes in the central nervous system (CNS).
Clinical presentation of hepatic encephalopathy
- Disturbance of sleep is often the earliest sign of hepatic encephalopathy.
- Asterixis and hyperreflexia.
- Fetor hepaticus (musty odor of breath).
- Alterations in personality and cognitive function.
Pathogenesis: Severe cortical vasoconstriction.
Clinical presentation of hepatorenal syndrome
- Signs and symptoms: Decreased glomerular filtration rate (GFR), oliguria, low urine sodium and disproportionately high ratio of blood urea nitrogen (BUN) to creatinine (i.e., prerenal pattern of acute renal failure).
- Clinical course: Often progressive and fatal, with mortality rate of 95%.
Important point: Kidneys are histologically normal.
Cirrhosis of the Liver
Basic description: Diffuse scarring of the liver with nodular regeneration of hepatocytes, resulting in severe disruption of hepatic architecture (Figure 15-4).

- Viral hepatitis; hepatitis C (about 50% of patients with hepatitis C infection develop cirrhosis). Hepatitis C has now surpassed alcoholism as the leading cause of cirrhosis in the United States. Hepatitis B is less likely to progress to cirrhosis.
- Alcohol abuse (6–15% of chronic alcoholics develop cirrhosis).
- Nonalcoholic fatty liver disease (NAFLD) or nonalcoholic steatohepatitis (NASH). Incidence is rising dramatically. Up to 10% of patients with NAFLD/NASH will go on to develop cirrhosis.
- Sclerosing cholangitis, primary biliary cirrhosis, biliary atresia, autoimmune hepatitis, hereditary hemochromatosis, Wilson disease, and α1-antitrypsin deficiency are less common causes of cirrhosis.
- Cryptogenic cirrhosis: Term used when there is no recognizable cause for the cirrhosis.
Source of Cirrhosis | Causes |
|---|---|
Infectious | Viral (hepatitis B, C, D) |
Toxins | Ethanol |
Metabolic | Hereditary hemochromatosis, Wilson disease, α1-antitrypsin deficiency |
Biliary disease | Primary biliary cirrhosis, primary sclerosing cholangitis |
Other | Autoimmune, cryptogenic |
Mechanism of cirrhosis: Chronic inflammation causes the release of transforming growth factor-β (TGF-β), which promotes collagen synthesis by stellate cells, leading to fibrosis. Cycles of cell death, fibrosis, and regeneration result in cirrhosis of the liver; reorganization of vascular microarchitecture plays a role.
Complications of cirrhosis include portal hypertension, ascites, hepatocellular dysfunction, portal vein thrombosis, and hepatocellular carcinoma (Figure 15-5 A–D).
- Portal hypertension: The portal vein normally drains into the liver. If the liver is scarred, intrahepatic vascular tone increases, causing an increased resistance to portal venous flow and thus an increase in portal venous pressure. Portal hypertension forces blood returning to the heart to take alternate routes such as esophageal varices, hemorrhoids, and caput medusae (i.e., dilated veins in abdominal wall). Portal hypertension contributes to the development of ascites and splenomegaly. Splenomegaly also traps red blood cells and platelets. Note: The cause of portal hypertension can be prehepatic, intrahepatic, or posthepatic, depending upon the site of the pathology. Cirrhosis is an intrahepatic cause of portal hypertension.
- Rupture of esophageal varices can lead to massive gastrointestinal hemorrhage.
- Ascites: Occurs as a result of portal hypertension (increased hydrostatic pressure) and hypoalbuminemia (decreased osmotic pressure).
- Important points
- Ascites is detectable on physical examination (shifting dullness and fluid wave) at amounts of > 500 mL.
- Serum-ascites albumin gradient (SAAG) > 1.1 is diagnostic of portal hypertension.
- Ascites is detectable on physical examination (shifting dullness and fluid wave) at amounts of > 500 mL.
- Spontaneous bacterial peritonitis
- Basic description: Infection of ascitic fluid, usually by Enterobacteriaceae or Pneumococcus.
- Symptoms and signs: Fever, abdominal pain, and tenderness; rebound tenderness and guarding; and leukocytosis.
- Outcomes: Can precipitate hepatic encephalopathy or renal insufficiency.
- Basic description: Infection of ascitic fluid, usually by Enterobacteriaceae or Pneumococcus.
- Important points
- Hepatocellular dysfunction, leading to impaired protein synthesis (e.g., decreased albumin and clotting factors), which causes bleeding tendencies and contributes to ascites. Hepatocellular dysfunction also leads to low BUN levels and elevated ammonia levels.
- Other complications of cirrhosis: Portal vein thrombosis, hepatocellular carcinoma, hyperestrinism leading to testicular atrophy and gynecomastia.
- Note: Remember, cirrhosis is one of the main causes of hepatic failure.
Figure 15-5.

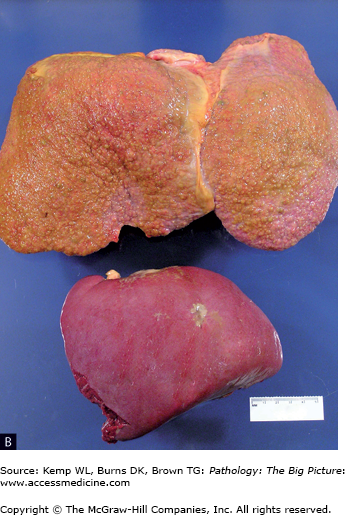

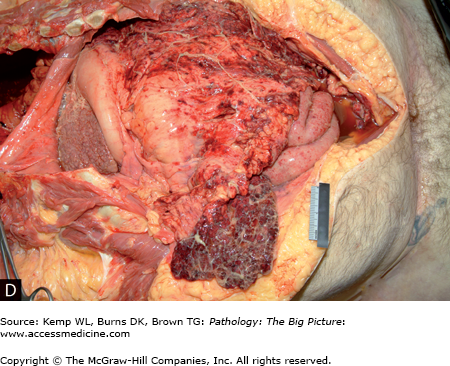
Complications of cirrhosis. A, Esophageal varices. The blue ring is attempted ligation of the varices by the patient’s physician. B, Congestive splenomegaly. With portal hypertension, blood return through the splenic vein is impaired. Normally, the spleen is about one tenth the size of the liver; in the photograph, the spleen is about one third the size of the cirrhotic liver. C, Ascites. Ascites occurs in patients with cirrhosis because of increased vascular pressure caused by portal hypertension, and also because of decreased plasma osmotic pressure caused by hypoalbuminemia. D, Spontaneous bacterial peritonitis. Patients with cirrhosis are prone to development of peritonitis, with no other underlying risk factors identified (e.g., perforated gastric ulcer, diverticulitis).
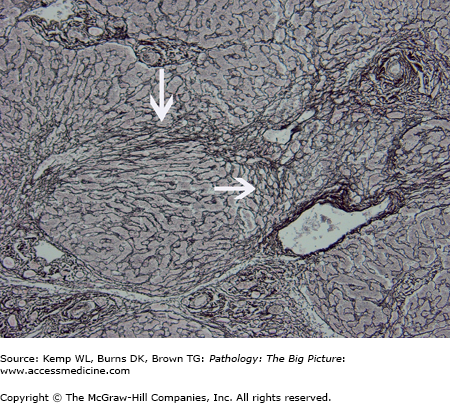
- Gross: Diffusely nodular liver, with a micronodular (< 3 mm nodules) or macronodular (> 3 mm nodules) architecture. Macronodular cirrhosis is associated with hepatitis, autoimmune diseases, and end-stage cirrhosis of all etiologies. Micronodular cirrhosis is associated with alcohol use and with other causes such as α1-antitrypsin deficiency and hemochromatosis.
- Microscopic: Bridging fibrosis between the portal tracts and central veins, which divide residual and regenerating hepatocytes into nodules. Other features depend upon the cause of cirrhosis.
Acute and Chronic Hepatitis
Overview: Acute and chronic hepatitis are nonspecific terms relating to the time course of the disease within the liver. The basic description of acute and chronic hepatitis, causes, clinical presentation, and important distinguishing points are discussed below.
Basic description: Inflammation of the liver lasting < 6 months.
Causes of acute hepatitis
- Viral: Hepatitis A, B, C, D, and E, Epstein-Barr virus (EBV), and cytomegalovirus (CMV).
- Toxins: Alcohol, acetaminophen, isoniazid, halothane, and phenytoin.
Clinical presentation
- Signs and symptoms: Enlarged and tender liver; AST and ALT increased 20–100 times normal levels.
- Results of acute hepatitis: Vary from complete resolution to rapid progression, with extensive necrosis and fatal outcome.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


