Overview
Diseases of the immune system take many forms, including hypersensitivity reactions, autoimmune disorders, and immunodeficiency states. Hypersensitivity reactions occur as one of four types (types I–IV). Autoimmune diseases are the result of a failure in the immune system to recognize self-antigens, resulting in production of antibodies that react against normal components of cells. Most of the autoimmune diseases are associated with one or more specific antibodies, which can be identified by laboratory tests to aid in diagnosis. Immunodeficiency states can be hereditary or acquired. A major cause of acquired immunodeficiency is human immunodeficiency virus (HIV) infection. The concepts of immunity are also important in regard to transplantation efforts. This chapter will discuss hypersensitivity reactions, transplantation pathology, autoimmune diseases, amyloidosis, and both hereditary and acquired immunodeficiency.
Hypersensitivity Reactions
Overview: There are four types of hypersensitivity reactions, each of which has a different mechanism. These four types of hypersensitivity reactions will be discussed below.
Mechanism: Exposure to an antigen results in the formation of IgE. The antigen reacts with CD4+ cells, which differentiate to TH2 cells. TH2 cells release interleukin-3 (IL-3), IL-4, and IL-5. IL-5 stimulates eosinophils, and IL-4 activates IgE-producing B cells. The IgE binds to mast cells. Subsequent exposure to the same antigen results in binding of the antigen to IgE bound to mast cells, with the consequence of degranulation of the mast cells and release of mediators (e.g., histamine). The release of mediators causes increased vascular permeability, leading to edema and increased smooth muscle contraction and eventually to bronchoconstriction.
Sequence of events in type I hypersensitivity reaction
Early phase (occurs within 5–30 minutes of exposure to antigen): Characterized by vasodilation, increased vascular permeability, and increased smooth muscle contraction. The early phase is due to binding of antigen to IgE bound to mast cells, with subsequent degranulation of the mast cells and release of mediators.
Late phase (occurs after 2–24 hours and lasts for days): Characterized by infiltration by neutrophils, eosinophils, basophils, and monocytes, and results in mucosal damage due to release of mediators by these recruited inflammatory cells.
Forms of type I hypersensitivity reactions
- Systemic anaphylaxis: Due to parenteral administration of antigen; for example, a bee sting or a reaction to penicillin.
- Local reaction: Urticaria (hives).
Causes: Penicillin, angiotensin-converting enzyme (ACE) inhibitors, intravenous (IV) contrast and other drugs, proteins (e.g., insect venoms), and food.
Clinical presentation of type I hypersensitivity reaction: Symptoms and signs include abrupt onset (within 30 minutes of exposure to antigen) of rash, nausea and vomiting and facial swelling, wheezing and stridor, and hypotension and tachycardia. Serum tryptase is a marker of anaphylaxis.
Complications of systemic anaphylaxis: Death due to airway compromise from laryngeal edema.
Overview of general mechanism: Antibodies directed against target antigens on cells or in extracellular matrix. The target antigens may be endogenous or absorbed exogenous antigens.
Specific mechanisms: There are three specific mechanisms by which type II hypersensitivity reactions occur. The three mechanisms are complement-dependent reactions, antibody-dependent cell-mediated cytotoxicity, and antibody-mediated cellular dysfunction.
- Complement-dependent reactions
- Mechanism: Antibody bound to antigen can fix complement and cause direct lysis of the cell through production of the membrane attack complex (MAC), or the complement can coat cells with C3b (an opsonin) and promote phagocytosis of the antigen.
- Example: Glomerulonephritis.
- Mechanism: Antibody bound to antigen can fix complement and cause direct lysis of the cell through production of the membrane attack complex (MAC), or the complement can coat cells with C3b (an opsonin) and promote phagocytosis of the antigen.
- Antibody-dependent cell-mediated cytotoxicity
- Mechanism: Cell types that bear receptors for the Fc portion of IgG, such as neutrophils, eosinophils, macrophages, and natural-killer (NK) cells, mediate removal of antigen.
- Examples: Transfusion reactions, erythroblastosis fetalis, and autoimmune hemolytic anemia.
- Mechanism: Cell types that bear receptors for the Fc portion of IgG, such as neutrophils, eosinophils, macrophages, and natural-killer (NK) cells, mediate removal of antigen.
- Antibody-mediated cellular dysfunction
- Mechanism: Antibodies themselves affect function of the antigen.
- Examples: Graves disease is due to an antibody that activates the thyroid-stimulating hormone (TSH) receptor, resulting in hyperthyroidism. Myasthenia gravis is due to antibodies against the acetylcholine (ACh) receptor, impairing neuromuscular transmission.
- Mechanism: Antibodies themselves affect function of the antigen.
Mechanism: Antibodies bind to the antigen, forming an immune complex. The antigens can be exogenous (e.g., viral proteins) or endogenous (e.g., DNA). These immune complexes can form in situ, or they can form in the vasculature and subsequently be deposited in organs, where they cause damage. The immune complex causes activation of the complement cascade. Note that immune complexes are commonly formed for various reasons, but only under certain circumstances do they elicit an immune reaction.
Examples: Immune–complex-mediated vasculitis and forms of glomerulonephritis.
General mechanism: Mediated by sensitized T cells rather than by antibodies.
Specific mechanisms
- Delayed form of type IV hypersensitivity reaction: CD4+ helper T cells (TH1 type) sensitized from previous exposure to an antigen secrete interferon-γ, which activates macrophages. Activated macrophages secrete IL-12, which causes differentiation of TH1 cells.
- Microscopic morphology: Stimulation of macrophages results in granulomas (i.e., collections of epithelioid histiocytes).
- Inciting agents: Mycobacteria, fungi, and parasites.
- Examples: Tuberculin reaction and contact dermatitis.
- Microscopic morphology: Stimulation of macrophages results in granulomas (i.e., collections of epithelioid histiocytes).
- Cell–mediated cytotoxicity: Sensitized CD8+ cells kill antigen-bearing cells. The antigens are presented by class I major histocompatibility complex (MHC) molecules. There are two mechanisms by which this occurs: the perforin-granzyme system and the FAS-FAS ligand system.
- Perforin–granzyme system: Perforin produces holes in the plasma membrane of cells, allowing granzyme to enter the cells. Granzyme then activates apoptosis through stimulation of caspase activity.
- FAS–FAS ligand system: The sensitized T lymphocytes have FAS ligand, which binds to FAS on target cells, leading to apoptosis.
- Perforin–granzyme system: Perforin produces holes in the plasma membrane of cells, allowing granzyme to enter the cells. Granzyme then activates apoptosis through stimulation of caspase activity.
Transplantation Pathology
Overview: Rejection of transplanted organs may be cellular or humoral, with cellular rejection mediated by T cells, and humoral rejection mediated by antibodies. In addition, rejection may be classified based upon its timing following the transplant procedure. The rejection can be hyperacute, acute, or chronic.
Mechanism: Cellular rejection is due to hypersensitivity of the recipient’s CD4+ cells, which results in killing of graft cells by CD8+ cells that have matured into cytotoxic T lymphocytes. The cytotoxic T lymphocytes kill graft cells through the perforin-granzyme pathway or the FAS-FAS ligand pathway.
Forms of cellular rejection
- Direct: The body recognizes MHC molecules on the surface of the antigen-presenting cells in the graft.
- Indirect: Antigens of the graft are presented by the recipient’s cells.
Overview: Humoral rejection is due to preformed antibodies or formation of antibodies against graft vasculature.
Hyperacute rejection
- Mechanism: Humoral reaction due to preformed antibodies to graft endothelium.
- Time course: Minutes following transplantation.
- Morphology: Grossly, there is cyanosis of the organ and a mottled parenchyma; microscopically, there is endothelial injury, neutrophils in arterioles, and infarcts of parenchyma.
Acute rejection
- Mechanism: Cellular or humoral reaction.
- Time course: Days to months to years following transplantation.
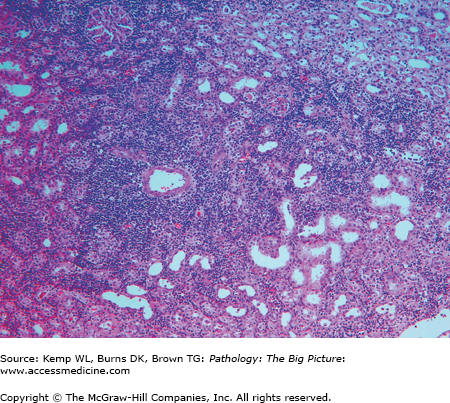
- Microscopic morphology of acute cellular rejection (Figure 3-1): Interstitial mononuclear infiltrate, edema, interstitial hemorrhage, and endothelialitis (i.e., swollen endothelial cells).
- Microscopic morphology of acute humoral rejection: Necrotizing vasculitis, neutrophilic infiltrate, and infarcts of parenchyma.
- Important point: An acute cellular rejection will respond to cyclosporine.
Chronic rejection
- Mechanism: Possibly, the indirect form of cellular rejection plays an important role.
- Time course: 4–6 months to years following the graft.
- Microscopic morphology: Vascular changes, interstitial fibrosis, interstitial mononuclear infiltrate, and ischemia with subsequent tissue loss.
Hematopoietic Transplantations
Basic description: Immune competent cells in the graft recognize antigens in the host.
Occurrence: In bone marrow transplants; in solid organ transplants when the organ is rich in lymphocytes (e.g., liver); and in non-irradiated blood.
Forms of GVHD
- Acute GVHD
- Time course: Days to weeks.
- Organs affected (and complications): Skin (rash), bile ducts (jaundice), and gastrointestinal mucosa (bloody diarrhea).
- Associated findings: Acute GVHD results in immunodeficiency and thus patients can have secondary infections, including cytomegalovirus (CMV) pneumonia.
- Time course: Days to weeks.
- Chronic GVHD
- Organs affected (and complications): Dermis and skin appendages (fibrosis), bile ducts (cholestatic jaundice), and esophagus (strictures).
- Associated findings: Chronic GVHD results in immunodeficiency; thus patients can have secondary infections, including CMV pneumonia.
- Organs affected (and complications): Dermis and skin appendages (fibrosis), bile ducts (cholestatic jaundice), and esophagus (strictures).
Autoimmune Diseases
Overview: Autoimmune disease results from a failure of self-tolerance. In self-tolerance, the body inactivates its immune response against antigens, which are present on and in its own cells. Autoimmune diseases can be organ specific or systemic, and are often associated with a specific antibody (Table 3-1).
Disease | Associated Antibodies |
|---|---|
Systemic lupus erythematosus | Anti-dsDNA, anti-Smith |
Drug-induced lupus | Antihistone |
Rheumatoid arthritis | IgM versus Fc portion of Ig |
Sjögren syndrome | Anti-SSA and anti-SSB |
CREST syndrome | Anti-centromere |
Diffuse scleroderma | Anti-scl70 |
General mechanism: Loss of self-tolerance. Contributing factors include susceptibility genes (e.g., certain HLA types such as B27 in ankylosing spondylitis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


