INTRODUCTION
The clinical management of kidney diseases is divided primarily between specialized internists (nephrologists) and specialized surgeons (urologists). Similarly, the pathologic evaluation of kidney diseases is divided between specialized pathologists (nephropathologists) who specialize in kidney diseases that are managed by nephrologists, and pathologists (urologic pathologists) who specialize in kidney diseases that are managed by urologists. Nephrologists and nephropathologists focus on so-called medical renal disease, which includes non-neoplastic diseases that affect the renal parenchyma. Urologists and urologic pathologists focus on diseases that can affect any level of the urinary system from kidneys to urethra, including congenital, infectious, and neoplastic diseases. This chapter will deal with the many forms of medical renal disease, and Chapter 13 with urologic kidney disease.
WHAT WE DOMedical Renal Disease Versus Urologic Disease and the Role of Nephropathologists
Nephrologists work closely with nephropathologists (renal pathologists) in the management of medical renal disease that requires renal biopsy. Many medical renal diseases, especially glomerular diseases, can only be definitively diagnosed by evaluating renal biopsy specimens. Further, repeat renal biopsies are important in the follow-up management of some patients to assess response to therapy, and to determine the degree of disease activity versus chronicity.
Kidney biopsies are most often obtained by percutaneous needle biopsy. The biopsy needle, usually with ultrasound guidance, is inserted into the patient’s flank. Because most medical renal diseases have diagnostic features only in the cortex, the goal is to obtain enough renal cortex for a definitive pathologic diagnosis. Renal biopsy specimens are routinely evaluated by light microscopy, immunofluorescence microscopy (or immunohistochemistry), and electron microscopy. Renal biopsies are the only major type of surgical pathology specimen that is routinely processed for electron microscopy. Immunofluorescence microscopy uses fluorochrome-labeled reagent antibodies to detect deposition of IgG, IgA, IgM, C3, C1q, kappa light chains, lambda light chains and fibrin in renal tissue, especially glomeruli. Special immunohistochemistry is required for some specimens, for example, to detect genetically determined protein abnormalities [e.g., abnormal glomerular basement membrane (GBM) collagen caused by mutations of collagen 4 genes], protein deposits other than immunoglobulin (e.g., amyloid A and myoglobin), and infectious pathogens (e.g., polyoma virus). Electron microscopy is able to detect ultrastructural changes that are not discernible by light microscopy but are essential for precise diagnosis. The importance of immunofluorescence microscopy and electron microscopy will become apparent as the diagnostic features of renal diseases are presented in this chapter.
GLOMERULAR AND VASCULAR DISEASES
QUICK REVIEWNormal Glomerular and Vascular Anatomy and Histology
The kidneys are highly vascularized organs with numerous vessels of many types that are the targets for multiple diseases.
At the hilum of the kidney, the main renal artery branches into anterior and posterior branches that feed into interlobar arteries that enter the renal parenchyma on either side of each papillary tip. The interlobar arteries feed into the arcuate arteries that run between the outer medulla and the cortex and give rise to interlobular arteries that run perpendicularly toward the renal capsule and give off multiple arterioles that become the afferent arterioles that enter glomerular tufts. Blood exits glomeruli via the efferent arterioles and subsequently provides sustenance to cortical and medullary tubules via peritubular capillaries. Thus, whenever there is glomerular injury, secondary tubular injury may occur and can be a conspicuous histopathologic feature in addition to the primary glomerular lesions.
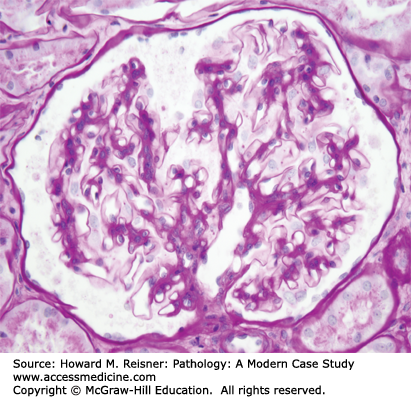
Glomerular disease accounts for a high proportion of medical renal diseases, and thus a clear understanding of glomerular structure and function is essential to understand many of the diseases discussed in this chapter. The glomerulus is a ball (Glomus) formed by interconnected capillaries (Figure 12-1). The glomerular capillaries (Figure 12-2) are supported by a stalk of mesangial cells that are highly specialized smooth muscle cells that have multiple functions in addition to providing support for the glomerular capillaries including regulation of capillary blood flow, and clearance of debris from the circulation and subendothelial zone. Glomerular capillaries are lined by specialized endothelial cells that are fenestrated, and thus allow relatively unimpeded access to the underlying GBM (Figure 12-2). Adjacent to the mesangium, the endothelial cell sits directly against the mesangial matrix. In the remainder of the capillary wall, the endothelial cell lies adjacent to the GBM.
GBM is produced primarily by the overlying epithelial cells (podocytes). The GBM surrounds the peripheral capillary wall and, at the juncture with the mesangium, splays out over the mesangium as the paramesangial GBM (Figure 12-2). Thus, the GBM does not completely surround the glomerular capillary lumen, which allows direct entry of materials from the plasma and the subendothelial zone into the mesangium.
The outer surface of the GBM is covered by podocytes that have foot processes that sit on the urinary space surface of the GBM (Figure 12-2). Extending between each foot process is a very thin slit diaphragm that resembles an adherens-like intercellular junction and provides the most selective barrier to the passage of proteins from the plasma into the urinary space. Integrity of the slit diaphragm appears to be the major determinant of capillary wall permeability to proteins.
GLOMERULAR DISEASE SYNDROMES
Injury to glomeruli causes a variety of clinical signs and symptoms because of disturbance of the many homeostatic functions of glomeruli. Disturbances in capillary wall integrity, especially injury to podocytes, result in increased permeability to proteins with resultant increased levels of protein in the urine (proteinuria). Injury to capillary walls that allows the cellular elements of blood to spill into the urine results in hematuria. Substantial impairment of blood flow through glomeruli, for example, caused by inflammation or scarring, results in reduced glomerular filtration that causes an elevation in blood urea nitrogen, increase in serum creatinine, and a reduction in glomerular filtration rate. Disturbance of blood pressure regulation and volume regulation results in hypertension.
Different forms of glomerular disease and different severities of glomerular injury tend to produce different combinations of signs and symptoms (Table 12-1). Patients with relatively mild glomerular disease may have asymptomatic proteinuria or asymptomatic hematuria or both, which is detected only by urinalysis. Diseases that cause severe proteinuria (greater than 3 g/24 hours) result in the nephrotic syndrome. Diseases that cause overt glomerular inflammation (glomerulonephritis) present with prominent hematuria often accompanied by hypertension, renal insufficiency, and proteinuria that usually are less severe than in patients with nephrotic syndrome. If glomerulonephritis results in a rapid loss of renal function, for example, requiring dialysis within 1 month, the disease is referred to as rapidly progressive glomerulonephritis (RPGN). Glomerular diseases may progress to chronic kidney disease, and, in some instances, to end-stage kidney disease. In the United States, diabetic nephropathy is the leading cause of end-stage kidney disease, hypertensive arterionephrosclerosis is the second leading cause, and all other forms of glomerular disease are the third leading cause. As will be discussed later in this chapter, glomerular injury is a major component of diabetic nephropathy and of hypertensive nephropathy.
| Nephrotic Syndrome | Nephritic Syndrome | |
|---|---|---|
| Edema | + to +++ | o to ++ |
| Proteinuria | ++ to +++ | + to ++ |
| Hypoproteinemia | + to +++ | o to + |
| Hematuria | o to ++ | ++ to +++ |
| Hyperlipidemia | ++ to +++ | o to + |
| Azotemia | o to ++ | o to +++ |
| Hypertension | o to + | o to +++ |
The nephrotic syndrome results from severe proteinuria (>3g/24 hours) with resultant edema, hypoproteinemia including hypoalbuminemia and hyperlipidemia including hypertriglyceridemia (Table 12-1). The edema of nephrotic syndrome is most often found in a periorbital and ankle distribution, but the edema can be widespread and even associated with anasarca (severe and generalized edema). In addition to proteinuria, patients with nephrotic syndrome often have lipiduria with lipids and lipoproteins in the urine, sometimes configured as oval fat bodies or fatty casts that can be seen by microscopic examination of urine. Mild proteinuria does not produce overt manifestations of nephrotic syndrome and is only detected by urinalysis. Although hematuria, hypertension, and renal insufficiency are more conspicuous features of the glomerulonephritic syndrome, all of these features may be present to some degree in patients with the nephrotic syndrome. Likewise, patients with a clear-cut glomerulonephritic syndrome with extensive hematuria, hypertension, and renal insufficiency virtually always have some degree of proteinuria and may have edema. The edema formation in glomerulonephritis is caused more by sodium retention, whereas the edema in nephrotic syndrome is caused primarily by reduced oncotic pressure in the plasma because of hypoproteinemia.
Some glomerular diseases most often present with nephrotic syndrome, whereas others most often present with glomerulonephritic (nephritic) syndrome and some tend to have overlapping features of both syndromes (Table 12-2). Glomerular diseases are categorized as primary when they are not associated with a systemic disease process or secondary when they are caused by (secondary to) a systemic disease. The primary glomerular diseases that most often present with the nephrotic syndrome include minimal-change glomerulopathy (minimal-change disease), membranous glomerulopathy, and focal segmental glomerulosclerosis (FSGS). Diabetic glomerulosclerosis and renal amyloidosis are example of secondary forms of glomerular disease that present with the nephrotic syndrome. Of the primary glomerular diseases that cause the nephrotic syndrome, minimal-change glomerulopathy is the most common cause of nephrotic syndrome in young children, membranous glomerulopathy is the most common cause of primary nephrotic syndrome in Caucasian adults, and FSGS is the most common cause of nephrotic syndrome in African-Americans.
| Nephrotic Syndrome | Asymptomatic Hematuria or Proteinuria | Acute or Recurring GN | Rapidly Progressive GN | |
|---|---|---|---|---|
| Minimal-change glomerulopathy | ++++ | o | o | o |
| FSGS | +++ | ++ | o | o |
| Membranous GN | +++ | + | ||
| MPGN/DDD | +++ | + | +++ | + |
| Alport syndrome | o | +++ | +++ | o |
| Thin GBM lesion | o | +++ | o | o |
| IgA nephropathy | + | +++ | +++ | + |
| Acute postinfectious GN | ++++ | o | o | |
| Lupus GN | + | +++ | ++ | |
| ANCA GN | + | ++ | ++++ | |
| Anti-GBM GN | o | + | ++++ |
CASE 12-1
A 35-year-old Caucasian male developed periorbital edema and swollen ankles. His primary care physician confirmed the edema. Blood pressure was normal. Point-of-care laboratory testing in the office revealed 4+ (out of 0-4+) protein and 1+ hematuria in the urine. He was referred to a nephrologists and further evaluation confirmed the edema and normal blood pressure. Laboratory findings included 4+ proteinuria with urine protein to creatinine ratio 4.7, 1+ hematuria with 5–10 dysmorphic red blood cells (RBCs)/hpf, 0–5 white blood cell (WBC)/hpf, serum creatinine 1.2 mg/dL, serum albumin 3.1 g/dL, serum cholesterol 355 mg/dL, and normal C3 and C4. Serologic testing for lupus, hepatitis B, and hepatitis C was negative.
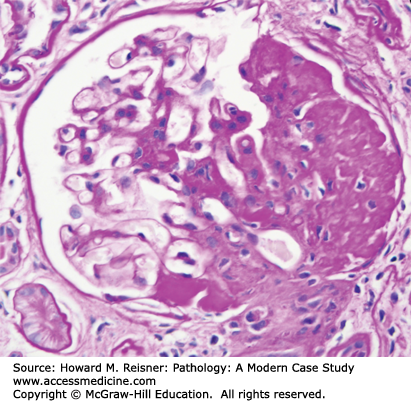
A renal biopsy was performed and was diagnostic for membranous glomerulopathy (Figures 12-3 and 12-4).
FIGURE 12-3
Membranous glomerulopathy with thick capillary walls but no hypercellularity by light microscopy (A. PAS stain, compare with Figure 12-1), and granular capillary wall staining for IgG by immunofluorescence microscopy (B).
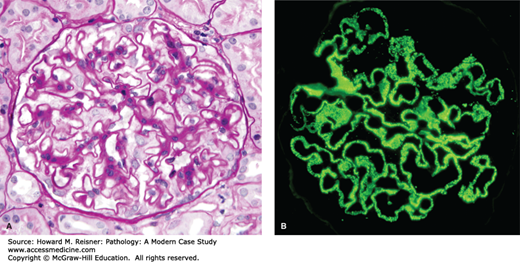
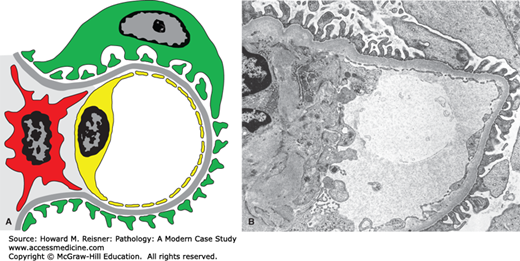
FIGURE 12-4
Diagram (A) and electron micrograph (B) of membranous glomerulopathy. Electron microscopic features of membranous glomerulopathy include numerous subepithelial electron-dense deposits that correspond to the immune complexes seen by immunofluorescence microscopy, and effacement of podocyte foot processes. Compare with Figure 12-2 (key as in Figure 12-2).

By light microscopy, membranous glomerulopathy is characterized by thickening of capillary walls in the absence of increased glomerular cellularity (hypercellularity) (Figure 12-3A). By electron microscopy, the capillary wall thickening is seen to be caused by the deposition of electron dense immune complex aggregates in the subepithelial zone between podocytes and GBM (Figure 12-4A and B). There may be deposition of varying amounts of GBM material between and around the deposits. There is effacement of podocyte foot processes overlying the deposits. Immunofluorescence microscopy demonstrates granular staining of glomerular capillaries with antibodies specific for immunoglobulin, especially IgG (Figure 12-3B), and complement, including C3. The subepithelial immune complexes form in situ in the subepithelial zone and are composed of antibodies from the circulation and antigens derived either from podocytes or from the circulation.
Approximately 75% of patients with membranous glomerulopathy have immune complexes composed of autoantibodies directed against antiphospholipase A2 receptor proteins that are produced by podocytes. The autoantibodies reach the subepithelial zone from the plasma and bind to antigens released by podocytes to form the subepithelial immune complex deposits. Membranous glomerulopathy also can be caused by (secondary to) antigens derived from systemic sources including infections (e.g., hepatitis B), cancer (e.g., carcinoma), and drugs (e.g., penicillamine). Membranous glomerulopathy also can be one of many phenotypes of glomerular injury caused by systemic lupus erythematosus. The nature of circulating autoantibodies and antigens in lupus determines whether a patient will develop a membranous glomerulopathy or a more inflammatory form of lupus glomerulonephritis that will be discussed later.
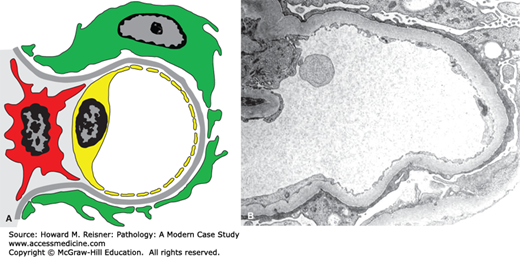
Minimal-change glomerulopathy (minimal-change disease) has no discernible histologic abnormalities by light microscopy, or only minimal increase in mesangial matrix or cellularity. By immunofluorescence microscopy, there is no staining for immunoglobulins or complement. The only consistent pathologic abnormality is seen by electron microscopy and is extensive effacement of podocyte foot processes (Figure 12-5). Instead of multiple individual foot processes normally lined up along the urinary surface of capillary walls, there is a more continuous layer of cytoplasm on the urinary surface of capillary walls. Even though the podocyte cytoplasm appears more continuous after foot process effacement than when the foot processes are intact, the podocytes are much more permeable to the passage of protein. Normally, there is a slit membrane between each podocyte that has low permeability to proteins, especially more positively charged larger proteins. In nephrotic syndrome, these adherens-like junctions between adjacent podocytes have increased permeability. Any form of glomerular disease with substantial proteinuria will have foot process effacement. However, in minimal-change glomerulopathy, the foot process effacement is the only pathologic lesion with none of the features of other glomerular diseases that are described in this chapter. Thus, the pathologic diagnosis of minimal-change glomerulopathy is a diagnosis of exclusion.
FIGURE 12-5
Diagram (A) and electron micrograph (B) of minimal-change glomerulopathy. Electron microscopic features of minimal-change glomerulopathy include effacement of podocyte foot processes in the absence of ultrastructural features of other diseases. Compare with Figure 12-2 (key as in Figure 12-2).
In the context of glomerular disease, focal indicates that less than 50% of glomeruli have a histologic abnormality, whereas diffuse indicates that 50% or more of glomeruli have a histologic abnormality. Segmental indicates that only a portion of a glomerular tuft has histologic injury, whereas global indicates that an entire glomerular tuft is injured. Thus, FSGS indicates that, at least at the beginning of the disease, only a portion of glomeruli have lesions by light microscopy and only a portion (segment) of the involved glomerular tufts have lesions. The term sclerosis indicates that there has been an accumulation of collagenous matrix at a site of injury. This is in essence a scar. In the setting of glomerular disease, the term sclerosis is used rather than fibrosis because the collagen is released by mesangial cells, endothelial cells, and podocytes rather than by fibroblasts.
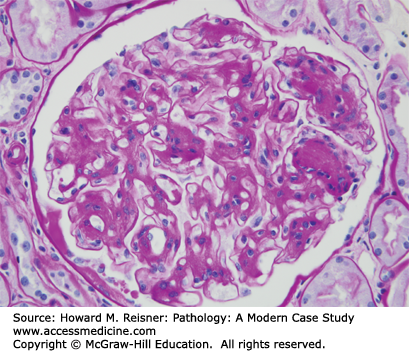
FSGS is characterized by focal segmental glomerular consolidation and sclerosis (scarring) (Figure 12-6). Many different forms of injury result in localized glomerular scarring. Therefore, to diagnose FSGS, other glomerular diseases that cause glomerular scarring must be ruled out, for example, focal segmental glomerulonephritis that has caused segmental scarring (sclerosis). By immunofluorescence microscopy, there is no evidence of immune complex deposition, and by electron microscopy, there is foot process effacement but no immune complex dense deposits. FSGS has many different etiologies and pathogenic mechanisms. As with other glomerular diseases, FSGS may occur as a primary (idiopathic) disease confined to the kidney, or can be secondary to a systemic process or a recognized etiology (Table 12-3).
|
FIGURE 12-6
Focal segmental glomerulosclerosis. Glomerulus with segmental sclerosis (PAS stain). Compare with Figure 12-1.
IN TRANSLATION Pathogenesis and Etiology of Focal Segmental Glomerulosclerosis
FSGS is pathologically, etiologically, pathogenetically, and clinically very heterogeneous (Table 12-3). Most FSGS is idiopathic with no known cause. However, in recent years a number of etiologies have been identified. The podocyte is the primary target of injury in some patients. This has been clearly demonstrated in patients with hereditary forms of FSGS who have mutations in genes that produce podocyte proteins, for example, nephrin, which is a major component of podocyte slit membranes. Overwork of glomeruli also can cause FSGS. One mechanism for overwork is compensatory hypertrophy in response to reduced renal tissue, for example, if one kidney and a portion of the other kidney are removed for cancer. The residual renal tissue undergoes hypertrophy and is at risk of development of focal FSGS. Fortunately, removal of one kidney is not sufficient to increase the risk of FSGS in the contralateral kidney, which allows living donors to provide kidneys for transplantation. Another mechanism for glomerular overwork is marked obesity, which results in compensatory glomerular hypertrophy and risk of development for FSGS. Not surprisingly, there is a marked increase in obesity-associated proteinuria and FSGS in parallel with the increasing incidence of obesity.
Diabetic glomerulosclerosis underlies the clinical syndrome of diabetic nephropathy, which is characterized by proteinuria, progressive decline in glomerular filtration rate and hypertension. Early mild diabetic glomerulosclerosis produces only very low levels of asymptomatic albuminuria (microalbuminuria). As diabetic glomerulosclerosis progresses, proteinuria becomes more severe and can reach nephrotic range. With further progression, there is loss of renal function.
The early pathologic lesion of diabetic glomerulosclerosis is thickening of GBMs and increase in mesangial matrix that can be so mild that it is only identifiable by electron microscopy (Figure 12-7). With progression, there is more and more mesangial matrix increase and thickening of GBMs. Eventually, the increase in mesangial matrix results in the formation of well-defined nodules called Kimmelstiel–Wilson nodules that can be seen by light microscopy (Figure 12-8). Similar glomerular changes occur in patients with type I and type II diabetes mellitus, although the glomerular disease in type II diabetes mellitus is more often complicated by changes of hypertensive injury in addition to the diabetic changes.
FIGURE 12-7
Diabetic glomerulosclerosis. Electron micrograph showing markedly thickened GBM and increased mesangial matrix. Compare with Figure 12-2B.
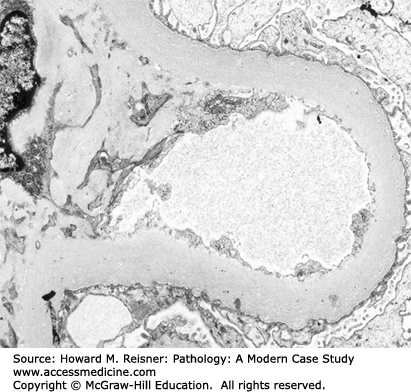
FIGURE 12-8
Diabetic glomerulosclerosis. Light microscopy showing segmental increase in mesangial matrix, including Kimmelstiel–Wilson nodules. Compare with Figure 12-1.
The clinical differential diagnosis in any patient with the nephrotic syndrome who has reached 60 years of age should include amyloidosis. Amyloidosis results from the accumulation of monotypic proteins in tissue in a unique configuration that results in the formation of randomly arranged fibrils. Different types of amyloid are caused by more than 20 different proteins. The two types of amyloid that most often cause glomerular disease are AL amyloid composed of immunoglobulin light chains and AA amyloid composed of amyloid A protein. In developed countries, most amyloid that causes kidney disease is AL amyloid, whereas, in developing or undeveloped countries, most amyloid that causes kidney disease is AA amyloid. The higher frequency of AA amyloidosis is the result of chronic inflammatory and infectious diseases that are more prevalent in these settings.
CASE 12-2
A 65-year-old Caucasian male developed periorbital edema and swollen ankles. His primary care physician confirmed the edema. Blood pressure was 155/97 mmHg. Point-of-care laboratory testing in the office revealed 4+ protein and 1+ hematuria in the urine. He was referred to a nephrologists and further evaluation confirmed the edema and hypertension. Laboratory findings included 4+ proteinuria with urine protein to creatinine ratio 4.9, trace hematuria with <5 dysmorphic RBCs/hpf, 5–10 WBC/hpf, serum creatinine 3.2 mg/dL, serum albumin 3.4 g/dL, serum cholesterol 320 mg/dL, and normal C3 and C4. Serum and urine protein electrophoresis and immunofixation revealed monoclonal IgG lambda. Serum-free lambda/kappa/ light chain ratio was markedly elevated. A bone marrow biopsy revealed slightly increased plasma cells but was not definitive for multiple myeloma.
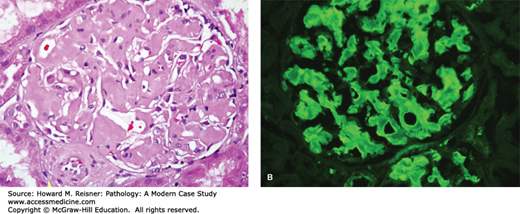
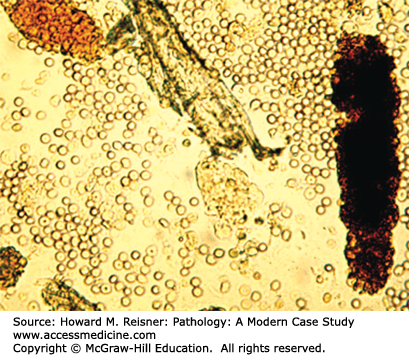
A renal biopsy demonstrated amyloid light chain (AL) amyloidosis composed of lambda light chains, but no light chain cast nephropathy (Figures 12-9 and 12-10).
FIGURE 12-9
AL amyloidosis. By light microscopy, amyloidosis causes replacement of normal glomerular architecture by amorphous acidophilic (eosinophilic) material (A, hematoxylin and eosin stain, H&E stain). Immunofluorescence microscopy of AL amyloid demonstrates deposits of monoclonal immunoglobulin light chains (B, anti-lambda).
IN TRANSLATION Monoclonal Immunoglobulin Pathogenesis
AL amyloid is caused by a B-cell dyscrasia that produces large amounts of monoclonal light chains resulting in amyloid deposits if the proteins have amyloidogenic biophysical properties. Lambda light chains cause AL amyloidosis more often than kappa light chains. Rare cases of amyloidosis are caused by abnormally truncated monoclonal heavy chains (AH amyloid). Not all monoclonal immunoglobulins have the same capability of causing kidney disease. As just noted, some monoclonal immunoglobulins cause AL amyloidosis with fibrillary deposits, whereas others cause a form of glomerular monoclonal immunoglobulin deposition with granular deposits (usually caused by kappa light chains), cast formation in tubular lumens resulting in acute tubular epithelial injury, accumulate in the cytoplasm of proximal tubular epithelial cells resulting in a tubulopathy, and are not associated with any kidney disease. There is evidence from experimental animal studies and tissue culture studies that different monoclonal immunoglobulins have different pathogenic capabilities based on as yet poorly understood differences in their biophysical characteristics. Because of this pathogenic variability, the differential diagnosis in a patient with kidney disease associated with monoclonal immunoglobulins in the blood or urine is broader than AL amyloidosis alone.
The glomerular diseases discussed thus far do not have overt inflammatory changes with an obvious influx of inflammatory cells. This is in contrast to glomerular diseases that usually present with more glomerulonephritic clinical features including prominent hematuria, and varying degrees of hypertension and renal insufficiency, and usually proteinuric of <3 g/day. The most characteristic feature of the glomerulonephritic (nephritic) syndrome is the presence of glomerular hematuria (Table 12-1). Asymptomatic hematuria without other clinical manifestations of glomerular disease tends to occur in mild or early glomerular disease.
More than 90% of all patients who have hematuria do not have glomerular hematuria. Most hematuria is the result of urinary tract disease and not glomerulonephritis. Common causes of urinary tract hematuria are urinary bladder infection, urethritis, prostatitis, urolithiasis, nephrolithiasis, and urinary tract neoplasms. Urinary tract hemorrhage other than glomerular hemorrhage is usually red or pink grossly, and microscopic examination during urinalysis demonstrates predominantly round RBCs often with a normal concave appearance. When it can be seen grossly, glomerular hematuria is tea-colored or coke-colored and by urinalysis there are many dysmorphic RBCs that have multiple blebs on the surface sometimes looking like Mickey Mouse ears. Another very distinct feature of glomerular hematuria is the presence of RBC casts in the urine that results from the accumulation of RBCs in the lumens of distal tubules where they become embedded Tamm–Horsfall protein to form a cylindrical cast that spills into the urine (Figure 12-10). The presence of these casts indicates that the hemorrhage has come from high up in the nephrons and not from the urothelial surface. If hematuria is caused by glomerulonephritis, there is almost always some degree of proteinuria, although this is usually less than nephrotic range (less than 3g/24 hours). Urologic hematuria is not accompanied by significant proteinuria. Depending on the nature of the underlying glomerular disease, glomerular hematuria may occur in the setting of
Asymptomatic hematuria
One episode of acute glomerulonephritis with or without acute renal failure (ARF)
Recurring episodes of acute glomerulonephritis
RPGN with rapid renal failure
Progressive chronic renal failure
Pathologically, inflammatory glomerular diseases (glomerulonephritides) typically have glomerular hypercellularity resulting from both influx of inflammatory cells, such as neutrophils, monocytes, and macrophages, and proliferation of glomerular cells, including mesangial cells, endothelial cells, and epithelial cells.
Pathologic diagnosis of glomerular disease required two components. One component is based on the pattern and severity of glomerular injury and is designated by descriptive terms such as mesangioproliferative glomerulonephritis, proliferative glomerulonephritis, necrotizing glomerulonephritis, and crescentic glomerulonephritis. The clinical presentation is influenced by the pattern of glomerular injury (Table 12-4). A second component of the diagnosis indicates the etiology or pathogenesis of the glomerular disease and includes terms such as IgA nephropathy, lupus glomerulonephritis, anti-GBM glomerulonephritis and antineutrophil cytoplasmic autoantibody (ANCA) glomerulonephritis. The etiologic and pathogenic category of glomerular disease correlates with the propensity to cause a certain pattern of glomerular injury, which in turn influences the likelihood of a particular clinical presentation (Table 12-2).
| Asymptomatic Hematuria | Acute or Recurring GN | Rapidly Progressive GN | |
|---|---|---|---|
| No lesion by light microscopy | ++++ | o | o |
| Mesangioproliferative GN | +++ | +++ | + |
| Proliferative GN | + | +++ | + |
| Crescentic GN | + | ++ | ++++ |
Systemic lupus erythematosus is an autoimmune disease that most often affects young African-American women but may occur in patients of any age or race. Virtually any organ of the body can be affected. The kidneys are one of the most frequent sites of injury and renal disease is a major cause of morbidity and mortality.
Lupus glomerulonephritis is caused by glomerular localization of immune complex deposits composed of autoantibodies of multiple specificities bound to the respective autoantigens and associated with activated complement components.
IN TRANSLATION Pathogenesis of Antibody-Mediated Glomerulonephritis
The inflammation that causes most forms of glomerulonephritis is mediated by antibodies. Antibody-mediated glomerulonephritis can be divided into
Immune complex-mediated glomerulonephritis
Antiglomerular basement membrane antibody (anti-GBM)-mediated glomerulonephritis
ANCA-mediated glomerulonephritis
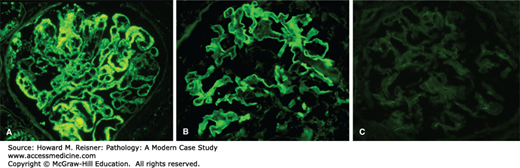
These categories can be distinguished by immunofluorescence microscopy (Figure 12-11), because
Immune complex-mediated glomerulonephritis has prominent granular deposits of immunoglobulin and complement in glomerular capillaries or mesangium or both (Figure 12-11A, left panel)
Anti-GBM disease has linear staining of GBMs for immunoglobulin (Figure 12-11B, middle panel)
ANCA-mediated glomerulonephritis has circulating ANCA with little or no immunoglobulin staining in glomeruli (Figure 12-11C, right panel)
Immune complexes that deposit or form in situ in glomeruli mediate inflammation by activating complement, and by activating leukocytes via Fc gamma receptor engagement. There are multiple categories of immune complex-mediated glomerulonephritis characterized by different locations and different compositions of the immune complexes, including membranoproliferative glomerulonephritis (MPGN), IgA nephropathy, postinfectious glomerulonephritis, lupus glomerulonephritis, and many others.
Anti-GBM antibodies bind directly to GBMs, in essence forming immune complexes in situ, and mediate inflammation by activating complement, and by activating leukocytes via Fc gamma receptor engagement.
ANCAs mediate inflammation by binding to target antigens (proteinase-3 or myeloperoxidase) that are displayed at the surface of circulating neutrophils causing them to adhere to small vessels (such as glomerular capillaries), undergo activation, and release destructive enzymes and oxygen radicals that injure vessel walls.
FIGURE 12-11
By immunofluorescence microscopy, immune complex-mediated glomerulonephritis has prominent granular deposits of immunoglobulin and complement in glomerular capillaries or mesangium or both (A, left panel), anti-GBM disease has linear staining of GBMs for immunoglobulin (B, middle panel), and ANCA-mediated glomerulonephritis has circulating ANCA with little or no immunoglobulin staining in glomeruli (C, right panel) (immunofluorescence with anti-IgG).
WHAT WE DO Pathologic Patterns of Glomerulonephritis: Proliferative, Necrotizing, Crescentic, and Sclerosing Glomerulonephritis
The pathologic diagnosis of glomerulonephritis includes both a descriptive term that characterizes the nature and severity of the histopathologic injury and a designation that indicates either the immunopathogenesis or etiology of the glomerulonephritis. The possible histopathologic patterns of glomerulonephritis are similar in all pathogenic categories; however, different pathogenic categories have different likelihoods of causing one type of histologic lesion versus another. The histopathologic patterns of injury in glomerulonephritis are determined by the severity of injury, the type of injury, and also the temporal stage of the disease with respect to acuity, chronicity, progression, and resolution.
As shown in Table 12-4, the clinical manifestations of glomerular disease correlate to a degree with the pattern of injury, and some categories of glomerulonephritis are more likely to produce particular patterns of glomerular injury rather than others. The crescentic glomerulonephritis category of injury that often is accompanied by RPGN clinically is important to recognize quickly and to diagnose accurately because timely and aggressive institution of immunosuppressive therapy is critically important for long-term preservation of renal function and optimum patient outcome.
No Lesion by Light Microscopy: With mild glomerular injury, immunofluorescence and electron microscopy may reveal a pathogenic process that is not detectable by light microscopy (i.e., glomeruli look normal by light microscopy). In this circumstance, patients usually have very mild clinical manifestations of glomerular disease such as asymptomatic hematuria, asymptomatic proteinuria or both.
Mesangioproliferative Glomerulonephritis (Figure 12-12A): Another relatively mild expression of glomerular injury is the presence of mesangial hypercellularity in the absence of other glomerular lesions. Mild mesangioproliferative glomerulonephritis may manifest with asymptomatic hematuria or proteinuria, or may have more overt clinical features of glomerulonephritis.
Proliferative Glomerulonephritis (Figure 12-12B, 12C): Overt inflammation of glomeruli results not only in mesangial hypercellularity but also proliferation of endothelial cells and influx of leukocytes including monocytes, macrophages, and neutrophils. This pattern of injury is referred to as endocapillary proliferative glomerulonephritis or simply proliferative glomerulonephritis. However, the term “proliferative” in this context should be understood to refer to hypercellularity caused not only by proliferative of glomerular cells but also to influx of leukocytes.
Crescentic Glomerulonephritis (Figure 12-12D):
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


