PRINCIPLES OF VIRAL DISEASES
The fundamental process of viral infection is the viral replicative cycle. The cellular response to that infection may range from no apparent effect to cytopathology with accompanying cell death to hyperplasia or cancer.
Viral disease is some harmful abnormality that results from viral infection of the host organism. Clinical disease in a host consists of overt signs and symptoms. A syndrome is a specific group of signs and symptoms. Viral infections that fail to produce any symptoms in the host are said to be inapparent (subclinical). In fact, most viral infections do not result in the production of disease (Figure 30-1).
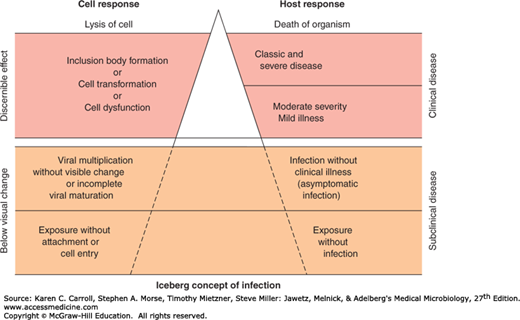
Important principles that pertain to viral disease include the following: (1) many viral infections are subclinical; (2) the same disease syndrome may be produced by a variety of viruses; (3) the same virus may produce a variety of diseases; and (4) the outcome in any particular case is determined by both viral and host factors and is influenced by the environmental context and genetics of each.
Viral pathogenesis is the process that occurs when a virus infects a cell and causes cellular changes. Disease pathogenesis is a subset of events during an infection that results in disease manifestation in the host. A virus is pathogenic for a particular host if it can infect and cause signs of disease in that host. A strain of a certain virus is more virulent than another strain if it commonly produces more severe disease in a susceptible host. Viral virulence in intact animals is not necessarily related to cytopathogenicity for cultured cells; viruses highly cytocidal in vitro may be harmless in vivo, and, conversely, noncytocidal viruses may cause severe disease.
Important features of two general categories of acute viral diseases (local, systemic) are compared in Table 30-1.
| Local Infections | Systemic Infections | |
|---|---|---|
| Specific disease example | Respiratory (rhinovirus) | Measles |
| Site of pathology | Portal of entry | Distant site |
| Incubation period | Relatively short | Relatively long |
| Viremia | Absent | Present |
| Duration of immunity | Variable—may be short | Usually lifelong |
| Role of secretory antibody (IgA) in resistance | Usually important | Usually not important |
PATHOGENESIS OF VIRAL DISEASES
To produce disease, viruses must enter a host, come in contact with susceptible cells, replicate, and produce cellular injury. Understanding mechanisms of viral pathogenesis at the molecular level is necessary to design effective antiviral strategies. Much of our knowledge of viral pathogenesis is based on cell culture and animal models because such systems can be readily manipulated and studied.
Specific steps involved in viral pathogenesis are the following: viral entry into the host, primary viral replication, viral spread, cellular injury, host immune response, viral clearance or establishment of persistent infection, and viral shedding.
Most viral infections are initiated when viruses attach and enter cells of one of the body surfaces—skin, respiratory tract, gastrointestinal tract, urogenital tract, or conjunctiva. The majority of these enter their hosts through the mucosa of the respiratory or gastrointestinal tract (Table 30-2). However, some viruses can be introduced directly into tissues or the bloodstream through skin wounds, needles (eg, hepatitis B and C, human immunodeficiency virus [HIV]), blood transfusions, or insect vectors (arboviruses).
| Route of Entry | Virus Group | Produce Local Symptoms at Portal of Entry | Produce Generalized Infection Plus Specific Organ Disease |
|---|---|---|---|
| Respiratory tract | Parvovirus | B19 | |
| Adenovirus | Most types | ||
| Herpesvirus | Epstein-Barr virus, herpes simplex virus | Varicella-zoster virus | |
| Poxvirus | Smallpox virus | ||
| Picornavirus | Rhinoviruses | Some enteroviruses | |
| Togavirus | Rubella virus | ||
| Coronavirus | Most types | ||
| Orthomyxovirus | Influenza virus | ||
| Paramyxovirus | Parainfluenza viruses, respiratory syncytial virus | Mumps virus, measles virus | |
| Mouth, intestinal tract | Adenovirus | Types 40 and 41 | |
| Calicivirus | Noroviruses | ||
| Herpesvirus | Epstein-Barr virus, herpes simplex virus | Cytomegalovirus | |
| Picornavirus | Some enteroviruses, including poliovirus, and hepatitis A virus | ||
| Reovirus | Rotaviruses | ||
| Skin | |||
| Mild trauma | Papillomavirus | Most types | |
| Herpesvirus | Herpes simplex virus | ||
| Poxvirus | Molluscum contagiosum virus, orf virus | ||
| Injection | Hepadnavirus | Hepatitis B | |
| Herpesvirus | Epstein-Barr virus, cytomegalovirus | ||
| Retrovirus | Human immunodeficiency virus | ||
| Bites | Togavirus | Many species, including eastern equine encephalitis virus | |
| Flavivirus | Many species, including yellow fever virus | ||
| Rhabdovirus | Rabies virus |
After entry, the viral nucleic acid and virion-associated proteins interact with cellular macromolecules to ultimately produce new virions that are released from the host cell by shedding or cell lysis. The specific mechanisms of viral replication are highly variable and can be quite complex, relying on one or more intermediate stages of production. The released virions are then able to attach and infect other cells in the immediate vicinity, causing local spread of infection.
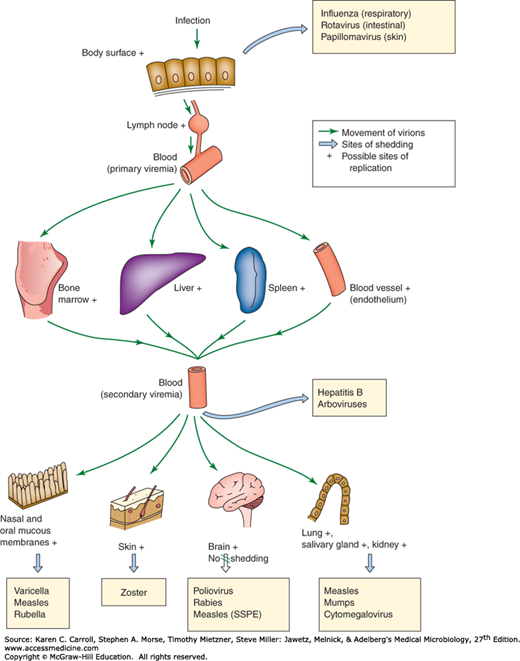
Some viruses, such as influenza viruses (respiratory infections) and noroviruses (gastrointestinal infections), produce disease at the portal of entry and typically do not spread systematically. Others can spread to distant sites (eg, cytomegalovirus [CMV], HIV, rabies virus) and cause additional disease manifestations (Figure 30-2). Mechanisms of viral spread vary, but the most common route is via the bloodstream or lymphatics. The presence of virus in the blood is called viremia. Virions may be free in the plasma (eg, enteroviruses, togaviruses) or associated with particular cell types (eg, measles virus) (Table 30-3). Viruses may multiply within those cells (eg, Epstein-Barr virus [EBV] is lymphotrophic and can replicate within white blood cells as it spreads). Some viruses travel along neuronal axons to spread within the host (eg, rabies migrates to the brain, herpes simplex virus [HSV] travels to ganglia to produce latent infection).
FIGURE 30-2
Mechanisms of spread of virus through the body in human viral infections. + indicates possible sites of viral replication; large arrows indicate sites of shedding of virus, with illustrative examples of diseases in which that route of excretion is important. Transfer from blood is by transfusion with hepatitis B and by mosquito bite in certain arboviral infections. SSPE, subacute sclerosing panencephalitis. (Modified from Mims CA, White DO: Viral Pathogenesis and Immunology. Copyright © 1984 by Blackwell Science Ltd. With permission from Wiley.)
| Cell Type Associated | Examples | |
|---|---|---|
| DNA Viruses | RNA Viruses | |
| Lymphocytes | Epstein-Barr virus, cytomegalovirus, hepatitis B virus, JC virus, BK virus | Mumps, measles, rubella, human immunodeficiency virus |
| Monocytes-macrophages | Cytomegalovirus | Poliovirus, human immunodeficiency virus, measles virus |
| Neutrophils | Influenza virus | |
| Red blood cells | Parvovirus B19 | Colorado tick fever virus |
| None (free in plasma) | Togavirus, picornavirus | |
Viruses tend to exhibit organ and cell-type specificities, or viral tropism. Tropism determines the pattern of systemic illness produced during a viral infection. As an example, hepatitis B virus has a tropism for liver hepatocytes, and hepatitis is the primary disease caused by the virus.
Tissue and cellular tropism by a given virus usually reflect the presence of specific cell surface receptors for that virus. Receptors are components of the cell surface with which a region of the viral surface (capsid or envelope) can specifically interact and initiate infection. Receptors are cell constituents that function in normal cellular metabolism but also happen to have an affinity for a particular virus. The identity of the specific cellular receptor is known for some viruses but is unknown in many cases.
The level of cell surface receptor expression and posttranslational modifications affect the ability of viruses to infect various cell types. For example, influenza virus requires cellular proteases to cleave virally encoded hemagglutinin in order to enable viruses to infect new cells, and expression of a glycolytic enzyme (neuraminidase) to release newly formed virions. Multiple rounds of viral replication will not occur in tissues that do not express the appropriate proteins.
Destruction of virus-infected cells in the target tissues and physiologic alterations produced in the host by the tissue injury are partly responsible for the development of disease. Some tissues, such as intestinal epithelium, can rapidly regenerate and withstand extensive damage better than others, such as the brain. Some physiologic effects may result from nonlethal impairment of specialized functions of cells, such as loss of hormone production. Clinical illness from viral infection is the result of a complex series of events, and many of the factors that determine degree of illness are unknown. General symptoms associated with many viral infections, such as malaise and anorexia, may result from host response functions such as cytokine production. Clinical illness is an insensitive indicator of viral infection; inapparent infections by viruses are very common.
Following a viral infection, the host will succumb, recover, or establish a chronic infection. Recovery mechanisms include both innate and adaptive immune responses. Interferon (IFN) and other cytokines, humoral and cell-mediated immunity, and possibly other host defense factors are involved. The relative importance of each component differs with the virus and the disease.
The importance of host factors in influencing the outcome of viral infections is illustrated by an incident in the 1940s in which 45,000 military personnel were inoculated with yellow fever virus vaccine that was contaminated with hepatitis B virus. Although the personnel were presumably subjected to comparable exposures, clinical hepatitis occurred in only 2% (914 cases), and of those only 4% developed serious disease. The genetic basis of host susceptibility remains to be determined for most infections.
In acute infections, recovery is associated with viral clearance and viral-specific antibody production. Establishment of a chronic infection involves complex interplay between viral and host immune factors, and the virus may enter a life-long latent state, or subsequently reactivate and cause disease months to years later.
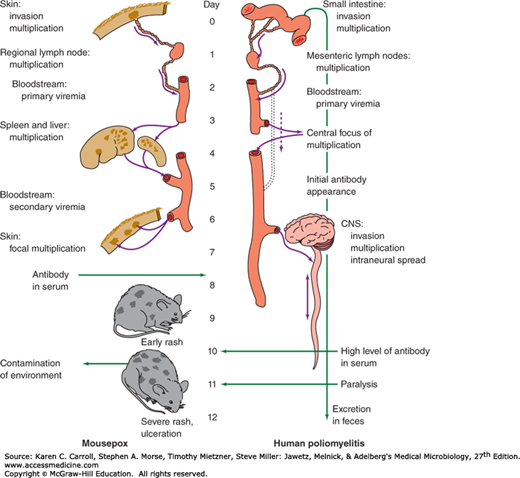
The last stage in pathogenesis is the shedding of infectious virus into the environment. This is a necessary step to maintain a viral infection in populations of hosts. Shedding usually occurs from the body surfaces involved in viral entry (see Figure 30-2). Shedding occurs at different stages of disease depending on the particular agent involved. During viral shedding, an infected individual is infectious to contacts. In some viral infections, such as rabies, humans represent dead-end infections, and shedding does not occur. Two examples of the pathogenesis caused by disseminated viral infections are shown in Figure 30-3.
FIGURE 30-3
Schematic illustrations of the pathogenesis of disseminated viral infections (mousepox and poliomyelitis). These viruses attach and replicate locally, spreading through the lymphatics and bloodstream to distant sites where they multiply further and can produce disease, followed by shedding into the environment from the initial site of infection. CNS, central nervous system. (Courtesy of F Fenner.)
The outcome of viral infections reflects the interplay between viral and host factors. Nonspecific host defense mechanisms are usually elicited very soon after viral infection. The most prominent among the innate immune responses is the induction of cytokines such as IFNs (see later discussion). These responses help inhibit viral growth during the time it takes to induce specific humoral and cell-mediated immunity.
The innate immune response is largely mediated by IFNs, which are host-coded proteins that are members of the large cytokine family that inhibit viral replication. They are produced very quickly (within hours) in response to viral infection or other inducers and are one of the body’s first responders in the defense against viral infection. IFNs also modulate humoral and cellular immunity and have broad cell growth regulatory activities.
There are multiple species of IFNs that fall into three general groups: designated IFN-α, IFN-β, and IFN-γ (Table 30-4). Both IFN-α and IFN-β are considered type I or viral IFNs; IFN-γ is type II or immune IFN. Infection with viruses is a potent inducer of IFN-α and IFN-β production; RNA viruses are stronger inducers of IFN than DNA viruses. IFNs also can be induced by double-stranded RNA and bacterial endotoxin. IFN-γ is not produced in response to most viruses but is induced by mitogen stimulation.
| Property | Type | ||
|---|---|---|---|
| Alpha | Beta | Gamma | |
| Current nomenclature | IFN-α | IFN-β | IFN-γ |
| Former designation | Leukocyte | Fibroblast | Immune interferon |
| Type designation | Type I | Type I | Type II |
| Number of genes that code for family | ≥20 | 1 | 1 |
| Principal cell source | Most cell types | Most cell types | Lymphocytes |
| Inducing agent | Viruses; dsRNA | Viruses; dsRNA | Mitogens |
| Stability at pH 2.0 | Stable | Stable | Labile |
| Glycosylated | No | Yes | Yes |
| Introns in genes | No | No | Yes |
| Homology with IFN-α | 80−95% | 30% | <10% |
| Chromosomal location of genes | 9 | 9 | 12 |
| Size of secreted protein (number of amino acids) | 165 | 166 | 143 |
| IFN receptor | IFNAR | IFNAR | IFNGR |
| Chromosomal location of IFN receptor genes | 21 | 21 | 6 |
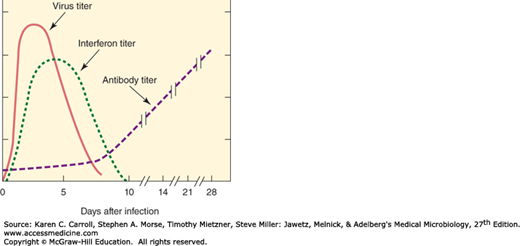
IFNs are detectable soon after viral infection in intact animals, and viral production then decreases (Figure 30-4). Antibody does not appear in the blood of the animal until several days after viral production has abated. This temporal relationship suggests that IFN plays a primary role in the nonspecific defense of the host against viral infections, as well as the fact that agammaglobulinemic individuals usually recover from primary viral infections about as well as normal people.
IFN is secreted and binds to cell receptors, where it induces an antiviral state by prompting the synthesis of other proteins that inhibit viral replication. Several pathways appear to be involved, including: (1) a dsRNA-dependent protein kinase, PKR, which phosphorylates and inactivates cellular initiation factor eIF-2 and thus prevents formation of the initiation complex needed for viral protein synthesis; (2) an oligonucleotide synthetase, 2-5A synthetase, which activates a cellular endonuclease, RNase L, which in turn degrades mRNA; (3) a phosphodiesterase, which inhibits peptide chain elongation; and (4) nitric oxide synthetase, which is induced by IFN-γ in macrophages.
Viruses display different mechanisms that block the inhibitory activities of IFNs on virus replication. Examples include specific viral proteins that block induction of expression of IFN (herpesvirus, papillomavirus, Filovirus, hepatitis C virus, rotavirus), block the activation of the key PKR protein kinase (adenovirus, herpesviruses), activate a cellular inhibitor of PKR (influenza, poliovirus), block IFN-induced signal transduction (adenovirus, herpesviruses, hepatitis B virus), or neutralize IFN-γ by acting as a soluble IFN receptor (myxoma virus).
Both humoral and cellular components of the adaptive immune response are involved in control of viral infections. Viruses elicit a tissue response different from the response to pathogenic bacteria. Whereas polymorphonuclear leukocytes form the principal cellular response to the acute inflammation caused by pyogenic bacteria, infiltration with mononuclear cells and lymphocytes characterizes the inflammatory reaction of uncomplicated viral lesions.
Virus-encoded proteins serve as targets for the immune response. Virus-infected cells may be lysed by cytotoxic T lymphocytes as a result of recognition of viral polypeptides on the cell surface. Humoral immunity protects the host against reinfection by the same virus. Neutralizing antibody directed against capsid proteins blocks the initiation of viral infection, presumably at the stage of attachment, entry, or uncoating. Secretory IgA antibody is important in protecting against infection by viruses through the respiratory or gastrointestinal tracts.
Special characteristics of certain viruses may have profound effects on the host’s immune response. Some viruses infect and damage cells of the immune system. The most dramatic example is the human retrovirus HIV that infects T lymphocytes and destroys their ability to function, leading to acquired immunodeficiency syndrome (AIDS) (see Chapter 44).
Viruses have evolved a variety of ways that serve to suppress or evade the host immune response and thus avoid being eradicated. Often, the viral proteins involved in modulating the host response are not essential for growth of the virus in tissue culture, and their properties are realized only in pathogenesis experiments in animals. In addition to infecting cells of the immune system and abrogating their function (HIV), they may infect neurons that express little or no class I major histocompatibility complex (MHC) (herpesvirus), or they may encode immunomodulatory proteins that inhibit MHC function (adenovirus, herpesvirus) or inhibit cytokine activity (poxvirus, measles virus). Viruses may mutate and change antigenic sites on virion proteins (influenza virus, HIV) or may downregulate the level of expression of viral cell surface proteins (herpesvirus). Virus-encoded microRNAs may target specific cellular transcripts and suppress proteins integral to the host innate immune response (polyomavirus, herpesvirus).
The immune response to one virus or vaccine may exacerbate the disease caused by subsequent infection with similar strains. For example, dengue virus hemorrhagic fever can develop in persons who already have had at least one prior infection with another dengue serotype due to the intense host response to infection.
Another potential adverse effect of the immune response is the development of autoantibodies through a process known as molecular mimicry
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


