Parkinson’s Disease
KEY CONCEPTS
![]() Thoughtful consideration of selection of initial therapy, management of drug dosing, and use of adjunctive therapies throughout the course of idiopathic Parkinson’s disease (PD) is necessary to optimize long-term therapeutic outcomes and minimize adverse effects.
Thoughtful consideration of selection of initial therapy, management of drug dosing, and use of adjunctive therapies throughout the course of idiopathic Parkinson’s disease (PD) is necessary to optimize long-term therapeutic outcomes and minimize adverse effects.
![]() The optimal time to start drug therapy in PD varies, but in general, treatment should be initiated when the disease begins to interfere with activities of daily living, employment, or quality of life.
The optimal time to start drug therapy in PD varies, but in general, treatment should be initiated when the disease begins to interfere with activities of daily living, employment, or quality of life.
![]() Surgery is reserved for patients who require additional symptomatic relief or control of motor complications despite receiving medically optimized therapy.
Surgery is reserved for patients who require additional symptomatic relief or control of motor complications despite receiving medically optimized therapy.
![]() Anticholinergic medication is useful for mild tremor-predominant PD but should be used with caution in the elderly and in those with preexisting cognitive difficulties.
Anticholinergic medication is useful for mild tremor-predominant PD but should be used with caution in the elderly and in those with preexisting cognitive difficulties.
![]() As monotherapy, amantadine and monoamine oxidase type B (MAO-B) inhibitors provide benefits in early PD, but the symptomatic effect is less than that of dopamine agonists and carbidopa/levodopa (L-dopa).
As monotherapy, amantadine and monoamine oxidase type B (MAO-B) inhibitors provide benefits in early PD, but the symptomatic effect is less than that of dopamine agonists and carbidopa/levodopa (L-dopa).
![]() Carbidopa/L-dopa is the most effective medication for symptomatic treatment, and eventually all patients with PD will require it.
Carbidopa/L-dopa is the most effective medication for symptomatic treatment, and eventually all patients with PD will require it.
![]() Most carbidopa/L-dopa–treated patients will develop motor complications (e.g., fluctuations and dyskinesias).
Most carbidopa/L-dopa–treated patients will develop motor complications (e.g., fluctuations and dyskinesias).
![]() MAO-B inhibitors and catechol-O-methyl-transferase inhibitors attenuate motor fluctuations in carbidopa/L-dopa–treated patients.
MAO-B inhibitors and catechol-O-methyl-transferase inhibitors attenuate motor fluctuations in carbidopa/L-dopa–treated patients.
![]() Dopamine agonists are effective and, compared to L-dopa, associated with less risk of developing motor complications but more risk to cause psychiatric symptoms, such as hallucinations and impulse control disorders.
Dopamine agonists are effective and, compared to L-dopa, associated with less risk of developing motor complications but more risk to cause psychiatric symptoms, such as hallucinations and impulse control disorders.
The presence of tremor at rest, rigidity, bradykinesia, and postural instability (instability of balance) are considered the hallmark motor features of idiopathic Parkinson’s disease (PD). These clinical features of PD were adeptly described in 1817 by James Parkinson.1
EPIDEMIOLOGY
Up to 1 million individuals in the United States have PD. The approximate annual incidence of PD (i.e., number of persons diagnosed with PD per year) is age-dependent and ranges from 10 per 100,000 persons in the sixth decade of life (i.e., 50 to 59 years of age) to 120 per 100,000 persons in the ninth decade of life (i.e., 80 to 89 years of age).2 Likewise, the prevalence of PD also increases with age, affecting 1% of people older than age 65 years and 2.5% of those older than age 80 years. The usual age at time of diagnosis ranges between 55 and 65 years. A higher incidence is reported among males, with a male-to-female ratio of up to 2:1.
ETIOLOGY
The true etiology of PD is unknown, but is likely the result of interactions between aging, genetic constitution, and environmental factors. In PD, a key histopathologic feature is degeneration of dopaminergic neurons in the substantia nigra that project to the striatum (i.e., the nigrostriatal pathway).3 Additionally, neuronal vulnerability in PD extends beyond the nigrostriatal pathway and includes specific neurons in autonomic ganglia, basal ganglia, spinal cord, and neocortex.4 In humans and primates, administration of the compound 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) results in a form of parkinsonism. The MPTP compound is converted by monoamine oxidase (MAO)-B to 1-methyl-4-phenylpyridinium ion (MPP+), a potent neurotoxin. MPP+ is toxic to neurons by inhibiting mitochondrial complex 1 of the electron transport chain, which results in the generation of excessive reactive oxygen species and cell death.5 Several synthetic pesticides have a molecular structure similar to that of MPTP. Although PD is sporadic, extensive epidemiologic research associates environmental factors, such as chronic exposure to pesticides, with an elevated risk for lifetime development of PD.6–8 Interestingly, epidemiologic studies have consistently associated an inverse correlation between cigarette smoking and caffeine consumption for development of PD.9,10
Intrinsically, the substantia nigra pars compacta (SNc) is a region characterized by high levels of oxidative stress because free radicals are generated from dopamine degradation (mediated by MAO; Figure 43-1). Several antioxidative molecules (e.g., glutathione) are present in the SNc to limit damage produced by free-radical reactions, but in PD, such protection might be overwhelmed or impaired. Thus, cellular damage from oxidant stress has long been discussed as an etiopathologic component of PD.11 The SNc is also rich in iron and copper, essential cofactors in the biosynthesis and metabolism of dopamine. The oxidation–reduction cycle of iron can also generate free radicals and toxic metabolites (Fig. 43-1). In addition, apoptosis (programmed cell death), excitotoxicity, inflammation, mitochondrial dysfunction, nitric oxide toxicity, proteosomal dysfunction, and autophagic cellular mechanisms are also implicated etiopathologic mechanisms in PD.
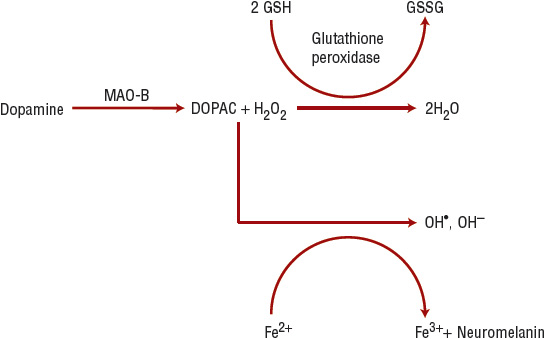
FIGURE 43-1 Dopamine metabolism results in hydrogen peroxide (H2O2) formation. If the glutathione system is deficient or excess hydrogen peroxide is present, hydrogen peroxide accepts an electron from ferrous iron (Fe2+), forming ferric iron (Fe3+), and the hydroxyl free radical (OH*). The hydroxyl free radical can cause lipid peroxidation, thereby damaging neuronal cell membranes. (DOPAC, 3,4-dihydroxyphenylacetic acid; GSH, glutathione; GSSG, glutathione disulfide; H2O, water; OH–, the hydroxide ion; MAO-B, monoamine oxidase B.)
Genetic susceptibility also plays a role. Although rare, several forms of familial parkinsonism have been linked to specific genetic mutations.12,13 For example, autosomal dominant forms of parkinsonism are associated with mutations of the α-synuclein: PARK1/PARK4 and leucine-rich repeat kinase 2 (LRRK:PARK8) gene loci. Autosomal recessive forms are associated with mutations of parkin:PARK2 and PTEN-induced putative kinase 1 (PINK1:PARK6) gene loci. Although less well defined, ongoing studies indicate that genetic polymorphisms also modify an individual’s risk for idiopathic PD.
PATHOPHYSIOLOGY
In the SNc, the two hallmark histopathologic features of PD are depigmentation of dopamine-producing neurons (i.e., loss of SNc neurons) and presence of Lewy bodies (neuronal cytoplasmic filamentous aggregates composed of the presynaptic protein α-synuclein) in the remaining SNc neurons. Lewy bodies appear in degenerating neurons in association with adjacent gliosis and the distribution of pathology is proposed to occur in stages.4 In the premotor stage of PD, Lewy bodies are initially found in the medulla oblongata, locus coeruleus, raphe nuclei, and olfactory bulb. This may correlate with observations that anxiety, depression, and impaired olfaction are detectable in premotor stages of PD. As PD progresses, Lewy pathology ascends to the midbrain (particularly the SNc) and accounts for development of motor features. In advanced stages, Lewy pathology spreads to the cortex, and this may correlate with cognitive and additional behavior changes. The observation that Lewy pathology can spread into adjacent healthy neurons has given rise to the postulate that prion-like propagation of α-synuclein aggregates may be occurring.
Pathologic findings reveal a correlation between the extent of nigrostriatal dopamine loss and the severity of certain PD motor features (e.g., bradykinesia). The threshold for onset of clinically detectable PD appears to be the loss of 70% to 80% of SNc neurons.14 Functional neuroimaging studies suggest compensatory responses, such as upregulation of dopamine synthesis and downregulation of synaptic dopamine reuptake, occur as adaptive mechanisms beginning in the premotor stage of PD. These adaptive responses may help to explain why the motor features are not clinically detectable until profound depletion (70% to 80%) of SNc neurons has occurred.
Dopaminergic projections from the SNc to the striatum (putamen and caudate) synapse on two major populations of dopamine receptor-mediated efferent neurons (referred to as the direct and indirect pathways), which, in turn, mediate motor activity via a complex neuronal circuit involving the extrapyramidal system (Fig. 43-2). In PD, the degeneration of the SNc neurons results in reduced activity within these two efferent pathways. The direct pathway involves activation of striatal dopamine1 (D1) dopamine receptors (which are coupled to adenylate cyclase) and stimulates the inhibitory γ-aminobutyric acid (GABA)/substance P efferents to the globus pallidus interna (GPi) and substantia nigra pars reticulata. The GPi and substantia nigra pars reticulata efferents are inhibitory to the thalamus.15 In PD, the reduced activation of D1 receptors results in greater inhibition of the thalamus. The indirect pathway involves activation of striatal dopamine2 (D2) dopamine receptors (which are coupled to a guanosine triphosphate-binding protein that opens potassium channels to hyperpolarize neurons, thereby reducing the excitability of the neuron).15 Activation of striatal D2 receptors inhibits GABA/enkephalin efferents (medium spiny neurons) to the globus pallidus externa. The globus pallidus externa projects GABA neurons to the subthalamic nucleus. Here, excitatory glutamatergic neurons project to the GPi. GPi output is inhibitory on the glutamatergic thalamic projections. In PD, the reduced activation of D2 receptors translates into greater inhibition of the thalamus. In PD, restoring activity at the D2 receptor appears to be of more importance than the D1 receptor for mediating clinical improvements. Overall, loss of the presynaptic nigrostriatal dopamine neurons in PD results in inhibition of thalamic activity and reduced activation of the motor cortex. Dopaminergic therapies help to restore motor activity.
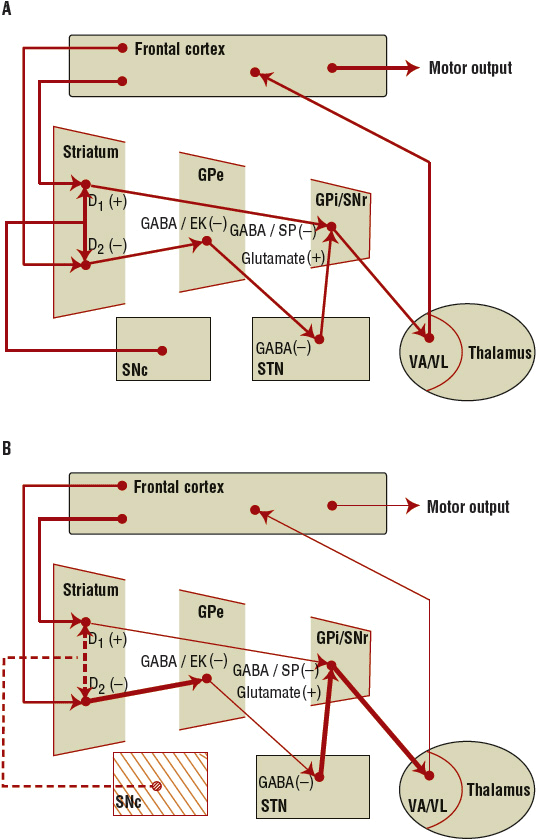
FIGURE 43-2 A. The normal balance of the basal ganglia–thalamocortical circuit. B. With nigrostriatal degeneration (dashed line), there is loss of inhibition of the GPi by the direct pathway and activation of the GPi via the indirect pathway, resulting in decreased activation of the cortex. See the text for details. (GPe, globus pallidus externa; GPi, globus pallidus interna; SNc, substantia nigra pars compacta; SNr, substantia nigra pars reticulata; STN, subthalamic nucleus; VA, ventroanterior nuclei of the thalamus; VL, ventrolateral nuclei of the thalamus.)
CLINICAL PRESENTATION Idiopathic Parkinson’s Disease
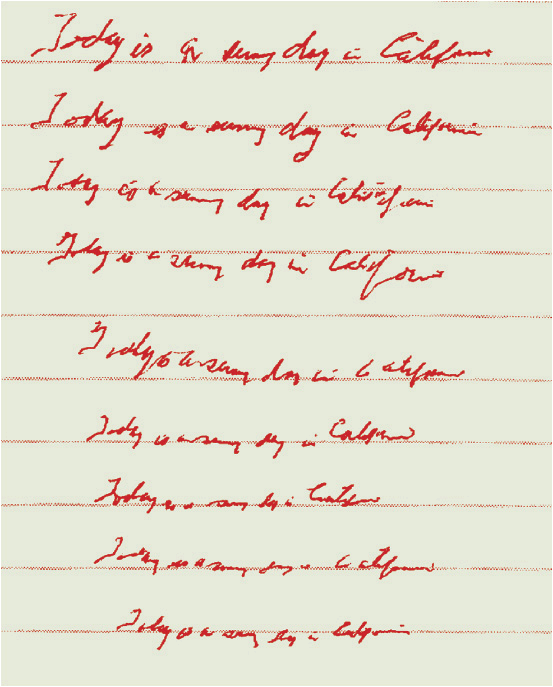
FIGURE 43-3 Example of micrographia in a patient with PD. As the sentence, “Today is a sunny day in California” is repeatedly handwritten, progressive diminution of letter size occurs (micrographia). The height of each lined row is approximately 5/16 inches (8 mm).(Courtesy of Jack J. Chen, PharmD, and David M. Swope, MD.)
In addition to dopamine, the synaptic organization of the basal ganglia also involves a variety of other neurotransmitters and neuromodulators, including acetylcholine, adenosine, enkephalins, GABA, glutamate, serotonin, and substance P. The potential role for drug modulation of these other neurotransmitters and receptor types is an active area of research and novel drug discovery for PD.16
Atypical parkinsonian disorders such as multiple system atrophy and progressive supranuclear palsy are characterized by damage to postsynaptic striatal neurons and dopamine receptors. Therefore, dopaminergic therapies provide less robust efficacy in atypical parkinsonism.
CLINICAL PRESENTATION
Although PD is unmistakable in its advanced form, recognizing PD during the early stages can be challenging. The clinical diagnosis of PD is based on the presence of bradykinesia and at least one of three other features: muscular rigidity, resting tremor, and postural instability (Table 43-1).17 Asymmetry of motor features is a supportive finding. It is important to note that tremor is not always present at the time of diagnosis, and postural instability typically occurs in later stages of PD. Overall, a diagnosis of PD can be made with a high level of confidence in a patient who has bradykinesia (along with rest tremor and/or rigidity), prominent asymmetry, and a good response to dopaminergic therapy. For the diagnosis of PD, other conditions must be reasonably excluded (Table 43-1). Medication-induced parkinsonism can mimic PD, so it is important to establish if such medications have been used (especially drugs that block D2 receptors, such as antipsychotics, metoclopramide, or phenothiazine antiemetics).18 Neurologic conditions that can be mistaken for PD include atypical parkinsonisms (e.g., corticobasal ganglionic degeneration, forms of multiple system atrophy, progressive supranuclear palsy) and essential tremor. Because the management and prognosis of PD differs from these other conditions, an accurate diagnosis is important. When the diagnosis is in doubt, referral to a movement disorders specialist is recommended.
TABLE 43-1 Diagnostic Criteria for Parkinson’s Disease and Differential Diagnosis

PD develops insidiously and progressively worsens. Over many years, symptoms can worsen to the point of severe disability, necessitating placement in a skilled nursing facility (especially with the development of dementia or frequent falling). However, the majority of patients remain community dwelling.
Tremor of an upper extremity occurring at rest (and occasionally an action or postural tremor) is often the sole presenting complaint; however, only two thirds of patients with PD have tremor on diagnosis, and some never develop this sign. Tremor in PD is present most commonly in the hands, sometimes with a characteristic pill-rolling motion. Less commonly, tremor may involve the jaw or legs. Like other motor features of PD, resting tremor often begins unilaterally and becomes bilateral with disease progression. Stressful or emotional (either negative or positive) situations often increase the tremor amplitude and severity. Usually, volitional movement abolishes resting tremor, and tremor is absent during sleep. Although resting tremor is visibly noticeable in PD and may cause social embarrassment for the patient, it often is the least physically disabling of the motor features.
Rigidity is the increased muscular resistance to passive range of motion and commonly affects the upper and lower extremities. If tremor is present in the affected extremity, the rigidity is associated with a cogwheel or ratchet-like quality upon examination. Facial muscles also are affected, resulting in hypomimia (masking of facial expressions) that may be erroneously interpreted as apathy, depression, or unfriendliness.
Bradykinesia refers to slowness of movement. Movement in PD is often slow throughout an intended action, and difficulty with the initiation of movement also occurs. A progressive slowing and decline in dexterity may impair tasks such as hand clapping, finger tapping, and handwriting (Fig. 43-3). Intermittent immobility (freezing) is another common characteristic. Freezing is especially likely to occur in situations such as when walking through a narrow doorway or initiating a turn. Patients also may experience a slow shuffling gait with difficulty halting their steps while in motion (festinating gait).
Postural instability, most common in advanced stages of PD, is one of the most disabling problems of PD because it increases the fall risk and is least amenable to pharmacotherapy. Testing for impaired postural responses by means of the pull test (in which a patient is unable to recover balance after sudden backward displacement at the shoulders) can help to identify the risk for falling. Many patients with impaired postural responses also have tendencies for propulsive gait (festination) and freezing, which also increases the risk of falling.
Although PD is known predominantly as a movement disorder, neuropsychiatric abnormalities also develop. Cognitive deterioration is not inevitable in PD; however, some patients deteriorate in a manner indistinguishable from Alzheimer’s disease and other dementing conditions.19 PD patients are also at increased risk for anxiety and depression.20 Although the disabilities of PD may provoke depression in some instances, the underlying biochemical changes in the brain associated with PD pathophysiology also predisposes for endogenous depression.
TREATMENT
Desired Outcomes
The goal in the management of PD is to improve motor and nonmotor symptoms so that patients are able to maintain the best possible quality of life.21 Specific objectives to consider when selecting an intervention include preservation of the ability to perform activities of daily living; improvement of mobility; minimization of adverse effects, treatment complications, putative disease modification; and improvement of nonmotor features such as cognitive impairment, depression, fatigue, and sleep disorders. To accomplish some of these objectives, consultation with a specialist is helpful (e.g., movement disorders, physical therapy, psychiatry, and sleep medicine).
General Approach to Treatment
![]()
![]() Once a correct diagnosis of PD is made, nonpharmacologic and pharmacologic interventions must be considered. Thoughtful consideration of selection of initial therapy, management of drug dosing, and use of adjunctive therapies throughout the course of PD is necessary to optimize long-term therapeutic outcomes and minimize adverse effects. The optimal time to start drug therapy in PD varies, but in general, treatment should be initiated when the disease begins to interfere with activities of daily living, employment, or quality of life. Figure 43-4 illustrates a general treatment approach for early and advanced PD, Table 43-2 summarizes antiparkinsonian medications and mechanisms of action, and Table 43-3 summarizes monitoring parameters for potential adverse effects. Treatment guidelines and monographs are updated frequently to keep up with new information and changes in treatment paradigms.22—26
Once a correct diagnosis of PD is made, nonpharmacologic and pharmacologic interventions must be considered. Thoughtful consideration of selection of initial therapy, management of drug dosing, and use of adjunctive therapies throughout the course of PD is necessary to optimize long-term therapeutic outcomes and minimize adverse effects. The optimal time to start drug therapy in PD varies, but in general, treatment should be initiated when the disease begins to interfere with activities of daily living, employment, or quality of life. Figure 43-4 illustrates a general treatment approach for early and advanced PD, Table 43-2 summarizes antiparkinsonian medications and mechanisms of action, and Table 43-3 summarizes monitoring parameters for potential adverse effects. Treatment guidelines and monographs are updated frequently to keep up with new information and changes in treatment paradigms.22—26
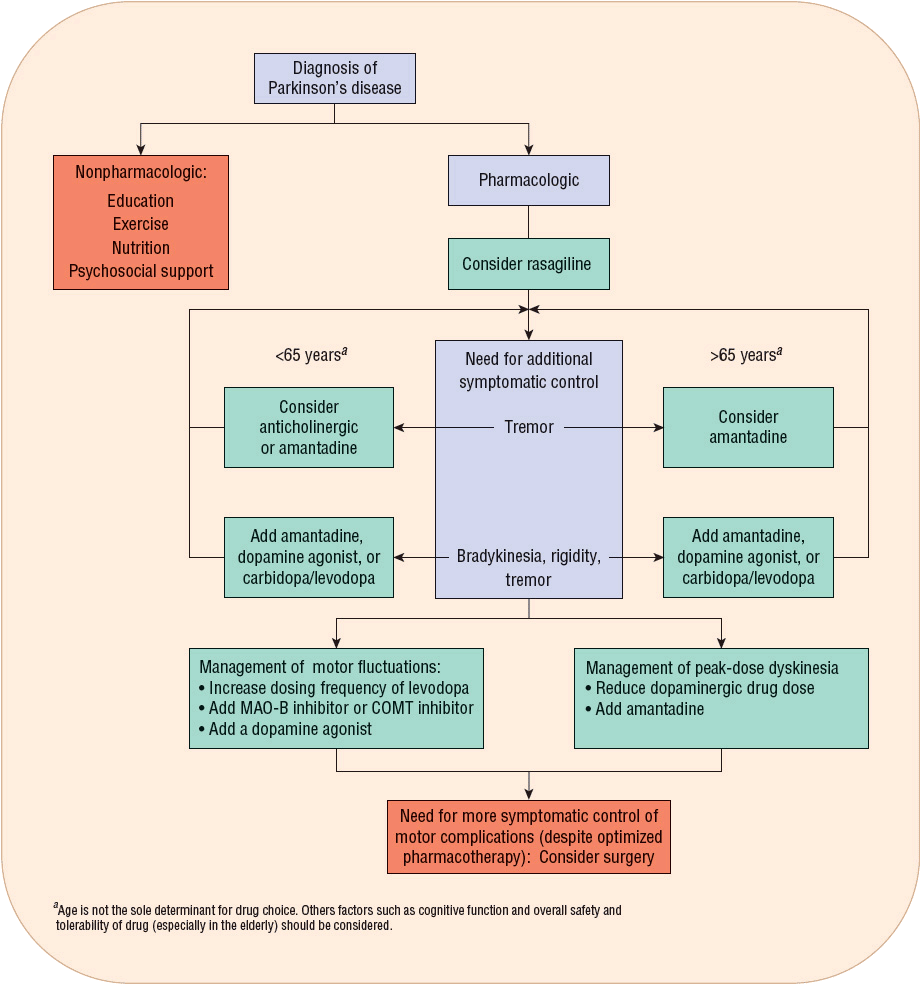
FIGURE 43-4 General approach to the management of early to advanced Parkinson’s disease.
TABLE 43-2 Dosing of Drugs Used in Parkinson’s Diseasea
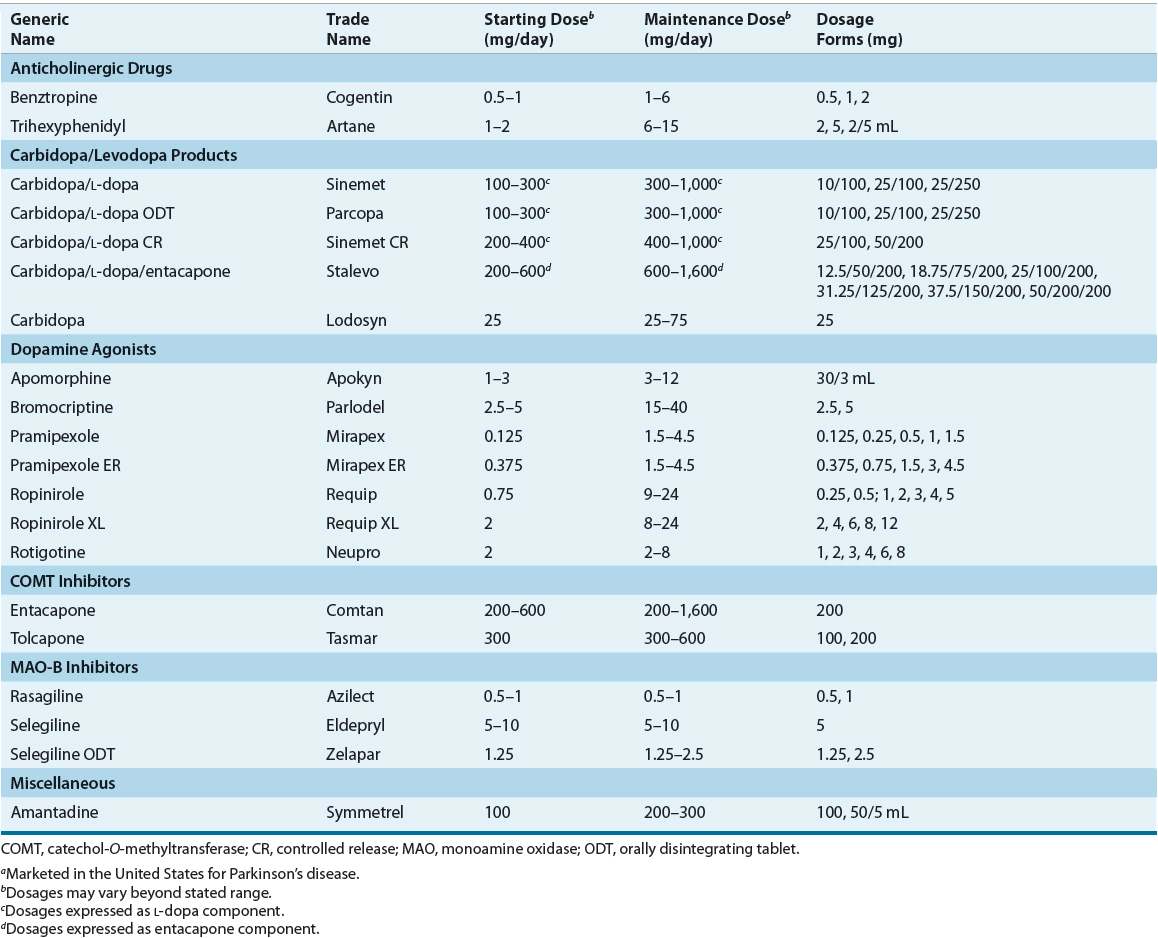
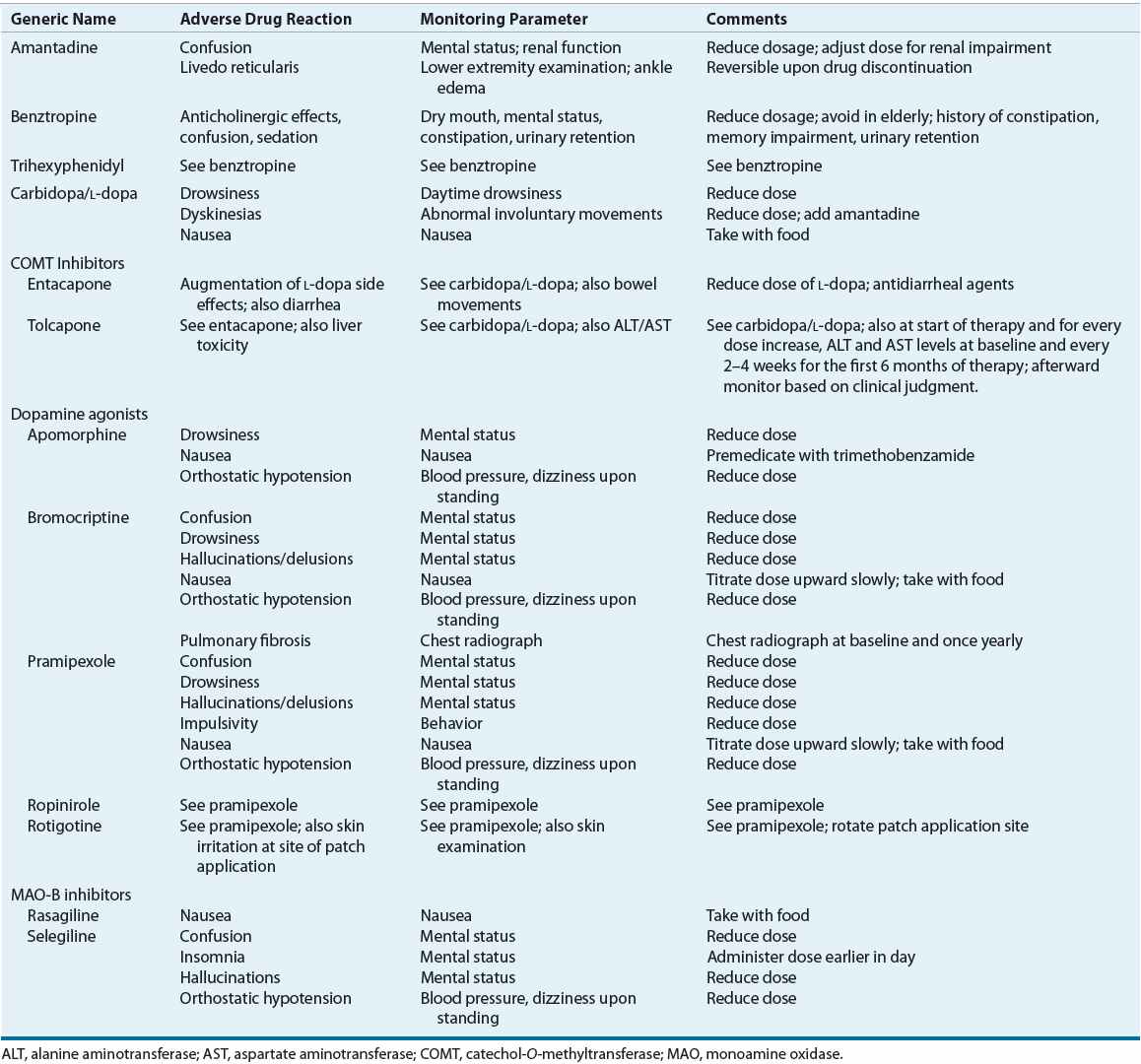
TABLE 43-3 Monitoring of Potential Adverse Reactions to Drug Therapy for Parkinson’s Disease
Clinical Controversy…
< div class='tao-gold-member'>



