Fig. 13.1
The most frequently used parenteral administration routes. Source: Recepteerkunde 2009, ©KNMP
Intravenous
Subcutaneous
Intracutaneous
Intramuscular
Intravenous injections are injected into the vein, so in the direction of the heart to diminish chance of bleeding on the puncture place (as would occur easily if intra-arterially would be injected). By this route the active substance is spread very fast through the circulatory system.
Subcutaneous injections are injected into the subcutaneous connective tissue of the upper arm, the upper leg, below or above the waist or the upper area of the buttock. Absorption of the active substances takes place through the vascular wall of the small vessels in the connective tissue. Heparins and insulins are usually injected subcutaneously.
Intracutaneous injection is done into the dermis layer of the skin, which is the tissue located under the epidermis. It is frequently done as a diagnostic measure, such as for tuberculin testing (screening test for tuberculosis referred to as a Tine test) and allergy testing (placing very small amounts of the suspected antigen or allergen in a solution under the skin).
Intramuscular injections are administered into the deltoid muscle of the upper arm or vastus lateralis muscles in the anterolateral aspect of the middle or upper thigh.
13.2.4 Specific Routes of Administration
Other more specific routes are employed for the administration of the active substance directly to the therapeutic site. For administration into the central nervous system the intrathecal, epidural or intracisternal injection route is used. Intrathecal injection is an injection into the spinal canal, more specifically into the sub-arachnoid space so that it reaches the cerebrospinal fluid and is useful in spinal anaesthesia, chemotherapy, or pain management applications. This route is also used for antibiotic treatment of infections, particularly post-neurosurgical. Medicines given intrathecally must not contain any preservative.
Since intravenous solutions have been accidentally applied into intrathecal space, international guidelines have been issued to prevent this occurring. A major recommendation is to use a non-luer connector (see Sect. 13.10.2) in neuraxial procedures. The first non-luer connectors for intrathecal administration were launched in UK. However, standardisation of these connectors is required [5].
Spinal anaesthesia (spinal block or sub-arachnoid block) is used to administer the injection into the subarachnoid space. Several local anaesthetics are used for spinal anaesthesia such as procaine, lidocaine, tetracaine, and bupivacaine. Vasoconstrictors such as adrenaline (0.1–0.2 mg) and phenylephrine (0.5–2 mg) can be added to subarachnoid blocks to decrease vascular uptake and prolong duration of action.
Local anaesthetics are synergistic with intrathecal opioids and intensify sensory block without increasing sympathetic block. The combination makes it possible to achieve spinal anaesthesia with otherwise inadequate doses of local anaesthetic.
The difference in density between the cerebrospinal fluid (CSF) and local anaesthetic solutions should be considered to restrict their distribution to the subarachnoid space. The relationship between the density of the local anaesthetic solution and the density of the CSF is called baricity. Solutions of anaesthetic substances, which have a greater density than CSF, are hyperbaric. Hyperbaric solutions are especially practical in small doses for unilateral anaesthesia. Several commercially available hyperbaric solutions contain 50–80 mg/mL glucose [6].
Epidural anaesthesia is a technique whereby a local anaesthetic is injected through a catheter placed into the epidural space, see Fig. 13.2.
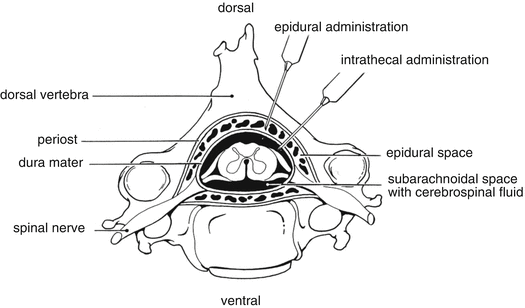
Fig. 13.2
Epidural and intrathecal administration route. Source: Recepteerkunde 2009, ©KNMP
Spinal and epidural techniques are shown to provide effective anaesthesia for caesarean section. Spinal block differs from an epidural block in a number of ways:
A smaller needle is used to perform a spinal block compared to an epidural block.
With the spinal technique the medicines are injected into the cerebrospinal fluid that bathes the spinal cord; with an epidural block, the medicines are delivered outside the dura, in the epidural space.
With the spinal technique small doses of local anaesthetic are required because they spread more easily in the spinal fluid.
A spinal block is a single injection of local anaesthetic medications and so there is only one opportunity to deliver the medications; in an epidural, an indwelling catheter may be placed that avails for additional injections.
Intraventricular administration is used for antineoplastic treatment of gliomas or delivering the therapeutic agents to different areas of central nervous system.
Intracisternal injection is used to deliver therapeutic agents to the cisterna magna.
Intraocular (or intravitreal) injections, containing active substances such as triamcinolone, antivascular endothelial growth factor (VEGF) inhibitors, antibiotics, antivirals, or antifungals, are administered into the eye.
By intraarticular injection medicines, e.g. containing corticosteroids, local anaesthetics as active substances, are administered into the joint.
In case of cardiac emergency medicines (e.g. adrenaline) are administered directly into the muscles of the heart.
More routes can be distinguished
Intracostal punction and insertion of a cannula into the pleural cavity is mostly used to induce pleurodesis and thereby reduce pleural effusions. Intrapleural administration of sclerosing agents such as sterile talcum powder, doxycycline or bleomycin destroy the mesothelial cell layer and incite pleuritis, adhesions, and destruction of the pleural space.
The arterial route is used to deliver some antineoplastics (e.g. cisplatin) to treat head and neck cancer.
Parenteral administration can also be done endotracheally. Through the tracheal tube medicine (containing e.g. atropine or epinephrine) is delivered directly into the patient’s bronchi.
13.2.5 Administration Methods
Parenterals can be administered by:
Direct (bolus) injection
Infusion by gravity
Injection/infusion by pump
During extracorporeal circulation.
Different types of veins can be punctured to be used for intravenous administration:
Small venous vessels (e.g. veins in the forearm)
Middle size venous vessel (e.g. vena brachialis)
Large central veins (e.g. vena jugularis or vena subclavia)
An infusion can also be administered subcutaneously. However the volume to be administered is limited to 20–30 mL per 24 h (see Sect. 13.10.3). During extracorporeal circulation procedures such as haemodialysis a parenteral liquid can be administered via ports in the dialysis devices.
13.3 Biopharmaceutics
The rate of absorption of the active substance and subsequent duration of action will be determined by:
Nature of the vehicle
Physico-chemical characteristics of the active substance
Interaction of active substance with vehicles, tissue and body fluid
13.3.1 Rapid Action
In emergency cases, e.g. cardiac arrest, anaphylactic shock or severe asthma, immediate action is required. Aqueous parenteral solutions administered intravenously are suitable for rapid onset of action.
13.3.2 Prolonged Action
Prolonged action of parenteral medicines is achieved by a different route of administration and different formulations. In general for example parenteral suspensions have a later onset and prolonged activity compared to parenteral solutions.
13.3.2.1 Intramuscular Administration
Administration into the muscle results in a delayed onset of action because the active substance has to be absorbed in the muscle, passed through capillary walls to be transported via the bloodstream to reach the site of action. Because most capillaries are fenestrated, all substances, whether lipid-soluble or not, cross the capillary wall at rates that are extremely rapid compared with their rates across other body membranes.
In addition the nature of the formulation affects the absorption rate of medicines administrated by intramuscular route.
Differences Between Muscles
The absorption rate after intramuscular administration differs depending on type of muscle chosen. Studies have shown that intramuscular injections result in different plasma concentrations of narcotics and perceived pain relief depending on the type of muscle used for administration. This was also found for the response to vaccination and use of antibiotics and insulin [7]. Absorption of active substances from the intramuscular site depends on the quantity and composition of the connective tissue and the rate of vascular perfusion of the area. Blood flow in the muscles varies (it is increased in deltoid muscle) and is influenced by the exercise of the muscle and morbidity. The muscles are covered with the subcutaneous connective tissue, a lipid layer (adipose layer) and the skin. The thickness and the lipid content of these tissues are different in different body areas. The subcutaneous fat layer at the gluteal intramuscular injection site is thicker in females (mostly > 3 cm) than in males. The medication should be administered with a needle long enough to reach the muscle without penetrating underlying structures.
13.4 Side Effects and Toxicity
Parenteral administration can be associated with pain and additional side effects and toxic reactions. These reactions can be more rapid and intense than with oral or cutaneous administration and may need fast intervention. Relevant types of adverse reactions are Protein hypersensitivity, (Thrombo)phlebitis, Pain, Extravasation and Damage by foreign particles.
Specific active substances exert their common adverse reactions when administered parenterally. Their rapid onset may lead to severe problems and therefore some substances require special attention beforehand:
Benzylpenicillin (potential allergic reaction).
Rituximab and other chimeric antibodies (infusion-related reactions such as hypotension, cardiopulmonary reactions, angioedema; mild to moderate infusion-related reactions such as fever, chills and rigors occur frequently).
Phenytoin (cardiovascular reactions, slow injection is associated with a high probability of precipitation).
13.4.1 Protein Hypersensitivity
With an increasing use of parenterally administered proteins there is an increased risk for hypersensitivity reactions. Proteins of any origin might elicit an immune response. Antibody formation against the protein may induce different results, e.g. no impact on efficacy, decrease of efficacy and neutralisation of the physiological protein. Reduced activity is derived from reduced half-life of the protein molecules in the circulation and rapid clearance by immune cells. Many factors influence the immunogenicity of proteins, including changes in the primary structure (sequence variation and glycosylation), storage conditions and changes in the tertiary structure (denaturation or aggregation) (see Sect. 18.4.1.4), contaminants or impurities arising from the production process, dose and length of treatment, the route of administration, and patient characteristics. Aggregation, which includes formation of dimers and high-order aggregates, may be mediated by pH, ionic strength, concentration, counter-ion composition of the formulation, temperature, sheer stress. Purification and concentration processes such as ultrafiltration, ion-exchange chromatography, lyophilisation, precipitation or ‘salting out’ can induce aggregation (see Sect. 22.2.5). Appropriate formulation of a protein product and stabilisation of the active substance is therefore extremely important. In any case the proteinaceous medicine should be stored and handled under optimal conditions. Freeze-dried particles should be completely dissolved, the recommended storage conditions should be adhered to, the preparation should not be shaken and should be not be administered with a peristaltic pump. The route of administration can influence the immunogenicity of the protein. Subcutaneous injection is associated with higher potential of immunogenicity than i.v. administration [8].
13.4.2 (Thrombo)phlebitis
Phlebitis is an infection of a blood vessel. Infusion-related phlebitis is characterised by pain, tenderness, erythema, induration, oedema and a local temperature increase. These circumstances may lead to thrombus formation or venous tissue destruction or both.
The incidence and duration of phlebitis seems to be dependent on a variety of factors. Physical-chemical factors such as low pH, hypertonicity, particles and precipitation play a role in the cause. Active substances that are poorly soluble in water may precipitate and can cause acute phlebitis. Active substances with adequate aqueous solubility may tend to cause phlebitis only because of prolonged or chronic administration. Clinical factors involving injection technique (infiltration, extravasation, type of needle, duration of infusion) but also irritating characteristics of the active substance can contribute to the occurrence of phlebitis [9, 10]. Sometimes (septic) phlebitis is caused by bacterial infection (e.g. cause of inappropriate aseptic technique during catheter insertion) and is characterised by inflammation with suppuration of the vein wall. Local responses to the parenteral challenges can be diminished by dilution of the medicine or by central venous instead of peripheral venous administration (see Sect. 13.10.3).
13.4.3 Pain
Parenteral administration can be associated with pain at the injection site. The so-called injection fear may be diminished by applying topically anaesthetics prior to injection. Eutectic mixtures of local anaesthetics (e.g. lidocaine/prilocaine cream or a tetracaine gel) have proven to be effective and well-tolerated in the relief of pain associated with intramuscular injections, venepuncture or intravenous injection in adults and children.
Non-physiological osmolality and pH or increased buffer concentrations in formulations can be responsible for pain at the injection site. Therefore some formulations for intramuscular use contain a local anaesthetic. Needle-free injection technologies are alternatives to reduce the intensity of injection pain [11].
13.4.4 Extravasation
Extravasation is the process by which any injection or infusion solution accidentally leaks into the surrounding tissue. The degree of damage due to extravasation depends on the active substance, the concentration, the localisation of the extravasation and the length of time an active substance may develop the damage. According to the type and severity of damage, active substances are categorised as irritants or vesicants [12].
13.4.5 Damage by Foreign Particles
The presence of visible foreign particles, which cannot be metabolised, must be avoided in injection solutions (see Sect. 32.12). However, the release of particulate matter (such as rubber chips, chemical fibres, glass fragments) is an intrinsic element of the production process. They originate from the formulation, processing equipment, primary package or the preparation process prior to use. Particles larger than 8 μm are generally trapped in the capillaries of the lungs, causing (thrombo) phlebitis and embolism. Smaller particles can be trapped in the liver, spleen and spinal cord [13]. Foreign body granulomas may result from intramuscular or subcutaneous injection of products containing foreign particles.
13.5 Product Formulation of Injections
13.5.1 Active Substance
The parenteral route is used for the administration of ‘small’ molecules as well as for ‘large’ molecules (proteins, monoclonal antibodies, vaccines, immunoglobulins).
13.5.2 Solubility of the Active Substance
Solubility can be improved by chemical modifications (change of pH, use of buffer, derivatisation, complexation, salt formation) and other techniques (use of excipients such as surfactants, solubilisers, cyclodextrines and phospholipids).
13.5.2.1 Buffers and pH Adjustment
For active substances that are ionisable, aqueous solubility can be raised by pH adjustment (see Sect. 19.1.1). An example for this principle is a quinine injection (Table 13.1). Quinine has low water solubility and a pKa around 4. By adjusting the pH of the quinine solution to pH 3, about 90 % of the quinine is ionised, resulting in a clear solution in the desired concentration.
Quinine hydrochloride | 12 g |
Hydrochloric acid | q.s. |
Water for injections | ad 100 mL |
13.5.2.2 Salt Formation
Salts of acidic and basic active substances have, in general, higher solubility than their corresponding acid or base forms. Haloperidol is almost insoluble in water (< 0.1 mg/mL) but haloperidol hydrochloride dissolves up to 3 mg/mL. Salt formation of haloperidol with lactic or tartaric acid results in increased water solubility and allows producing haloperidol injection solution (Haldol®) with a concentration up to 5 mg/mL.
On the other hand decreased solubility of an active substance and the formulation as a suspension may be advantageous. The duration of action of insulin is enhanced by forming salts with protamin and zinc with a lower solubility and lower rate of dissolution of insulin.
13.5.2.3 Complexation
Aqueous solubility of an active substance with low water solubility may be increased by molecular complexation with cyclodextrins, polyvidone and macrogols.
13.5.3 Vehicle
Water for injection is the preferred vehicle for the preparation of parenteral solutions (injections and infusions). When the active substance is poorly or not soluble in water, co-solvents or non-aqueous vehicles can be employed, or dosage forms such as liposomes or microspheres can be used.
13.5.3.1 Co-solvents
Co-solvents lower the surface tension of water resulting in increased solubility of poorly water soluble active substances. The most often used co-solvents in licensed injections are ethanol, glycerol, propylene glycol and macrogols (see Sects. 23.3.2, 23.3.3 and 23.3.4). The combination of propylene glycol and ethanol is commonly used to dissolve lipophilic active substances [15].
Diazepam is only slightly water-soluble and has a pKa of 3.4, which is not suitable to increase solubility by pH adjustment. The brand Diazepam-injection CF® 5 mg/mL contains diazepam solubilised in a co-solvent mixture of propylene glycol and ethanol.
Nimodipine is poorly soluble in water: < 0.1 mg/mL. The licensed pharmaceutical product Nimotop® infusion solution contains 0.2 mg/mL nimodipine and 170 mg/mL macrogol 400. This high concentration of macrogol is needed to dissolve the active substance but causes phlebitis when Nimotop® is administered via a peripheral vein. Therefore in the product information it is recommended to administer Nimotop® via a central venous catheter.
After diluting the infusion or injection concentrate with an aqueous solution or with blood the active substance may precipitate. This is due to the concomitant dilution of the co-solvent(s) and oversaturated solutions. Whether or not precipitation occurs in mixtures of water and co-solvents can be calculated in analogy to the calculation of the chance of precipitation in water, see Sect. 18.1.1.
Supersaturated solid solutions of poorly water-soluble compounds are inherently prone to recrystallisation over time (see Sect. 18.1.6). If the appropriate formulation principles and production processes are utilised, such systems can represent a formulation option for injection of poorly water-soluble active substances. However, crystallisation is not predictable. Serious clinical problems have been reported when precipitates have caused venous blockages [16].
Co-trimoxazole is the combination of trimethoprim (solubility 0.1–1 mg/mL; pKa = 7.4) and sulfamethoxazole (solubility < 0.1 mg/mL; pKa = 6). The licensed pharmaceutical preparation Bactrimel® is a concentrated co-trimoxazole solution of 16/80 mg/mL in propylene glycol, ethanol and water, with ethanolamine and sodium hydroxide. The pH of the solution is 9–10.
Before administration, the injection fluid 16/80 mg/mL has to be diluted to the concentration range of 0.125/0.625–8/40 mg/mL. The dilution causes reduced concentrations of organic solvents and a decreased pH value. Since the active substance precipitates slowly, the diluted preparation can be administered safely.
For an overview of the toxicity of co-solvents, when used parenterally, reference is made to the literature [17]. Glycerol is a safe co-solvent, but it is used infrequently in injectable formulations. It is often not able to achieve the desired concentration of the active substance. Ethanol improves the solubility, but is, in concentrations higher than 10 %, painful when injected. Propylene glycol elicits a haemolytic response in vitro and in vivo, which could be reduced by the addition of either saline or macrogol 400. Macrogol 300 and macrogol 400 are generally considered to be among the safest organic co-solvents. Although macrogols are biocompatible, peroxide impurities in macrogols can cause the degradation of oxidisable active substances [15].
13.5.3.2 Lipophilic Solvents
Vegetable oils such as cottonseed oil, peanut oil, and soybean oil can dissolve very lipophilic substances. Oils must be free from rancidity. Also semi synthetic lipophilic products such as isopropyl myristate and medium chain triglycerides are used in licensed parenteral liquids. They are stable and colourless.
Oily injectable formulations are only administered by intramuscular injection providing a depot for sustained drug delivery. A good example is the oil-based extended release injection product that contains haloperidol esterified to a decanoate dissolved in sesame oil. The volume to be injected is deposited deep into the gluteal muscle and forms a depot from where it is leached over a 1 month period into the blood stream according to its oil/water partition coefficient. This phenomenon together with the time needed for circulating enzymes to hydrolyse the ester into the active base is responsible for the prolonged length of action of this formulation.
Another example for a long-acting formulation is the intramuscular injection of fulvestrant, dissolved in castor oil and organic solvents. It is administered once monthly to treat hormone receptor positive breast cancer.
For intravenous or intramuscular administration, oil-soluble actives can be formulated as an oil–water-emulsion. When administered intravenously it is essential that the droplet size is about the same size as the lipid particles that circulate in the blood, the chylomicrons (0.2–3 μm). A typical emulsion contains 10–20 % soybean oil, 2 % glycerol and 1 % egg lecithin. These emulsions cannot be autoclaved. Coalescence of the droplets of the internal phase is a typical sign of instability. All-in-one total parenteral nutrition admixtures are a typical example for parenterally administered emulsion (compare Sect. 13.9).
Diprivan® or Disoprivan® is an example of a licensed medicinal product formulated as an emulsion. The active substance propofol (di-isopropyl phenol) is solubilised in an emulsion composed of 10 % soybean oil with egg phosphatide.
13.5.3.3 Microspheres and Liposomes
Microspheres and liposomes are described in Sect. 13.2.2 Some examples, described below, illustrate how these techniques are used to process substances with poor solubility into an injectable liquid.
DaunoXome®, used in treatment of Kaposi’s sarcoma, is an example of a liposomal antineoplastic medicine. Daunorubicin hydrochloride (1 mg/mL) is encapsulated in small liposomes. Amphotericin B, which is used in the treatment of systemic fungal infection, is also formulated as a stable colloidal particle.
The Swiss Serum Institute developed liposomal-based vaccines against hepatitis A and B, influenza, diphtheria and tetanus [18].
A liposomal injection solution of verteporin (Visudyne®) is formulated with egg phosphatidylglycerol and dimyristoylphosphatidyl choline. The freeze-dried powder, contains apart from the liposomes, verteporin, lactose as lyoprotector, and osmolality adjusting excipients. By reconstitution with water an opaque dark green injectable solution is generated, which is injected intravenously within the scope of photodynamic therapy.
Microsphere sizes range from 10 to 200 μm. The smaller the microspheres are, the better is their syringeability and the smaller are the needle diameters required for injection, which results in reduced patient discomfort. The larger the microspheres, the lower is the risk that the particles will be cleared from the injection site by macrophages. 10 μm is generally considered to be a safe lower size limit to avoid particle uptake by macrophages. The common microsphere size is about 30 μm. Microspheres can be administered at the site of action and guarantee prolonged activity by slow release of the active substance. Furthermore, sensitive active substances such as peptides and proteins may be protected from chemical and enzymatic degradation when entrapped in microspheres.
Several injectable prolonged release biodegradable microspheres are available as licensed products, e.g. Lupron Depot® and Risperdal® Consta [18, 19].The injectable ultrasound contrast agent (Optison®) consists of heat-denaturated albumin microspheres filled with the gas octafluoropropane. In a similar manner sulphur hexafluoride containing microspheres (SonoVue®) are used as injectable contrast agent in echocardiography.
13.5.3.4 Surfactants
Surfactants reduce surface tension and increase the solubility of lipophilic active substances in aqueous medium; solubilisates are formed (see Sect. 18.3.3). Lecithin, various phospholipids, polysorbate (Tween®) and polyoxyethylene castor oils such as Cremophor® EL can be used in injections [15].
Phytomenadione (Vitamin K1) appears as a non-ionisable, water-insoluble viscous liquid. In Konakion® MM injection fluid 10 mg/mL it is solubilised with lecithin and glycocholic acid creating micelles.
Amiodarone hydrochloride, which has a poor water solubility of 0.7 mg/mL, is solubilised in licensed pharmaceutical preparations thereby achieving a concentration of 50 mg/mL. Infusion solutions prepared with 5 % glucose as a vehicle still have amiodarone concentrations (6–15 mg/mL) exceeding by far the maximum concentration of water solubility. The resulting infusion solution contains the active substance still solubilised.
Polysorbate (Tween® 20 and 80) and Cremophor RH40® (macrogol glycerol hydroxyl stearate) or Cremophor EL® (polyoxylated 35 castor oil) can cause serious hypersensitivity reactions when administered parenterally [15]. They may cause incompatibilities by leaching plasticisers (such as phthalates) from polyvinyl chloride infusion devices.
The combination of 200 mg/mL polyoxyethylene hydrogenated castor oil and ethanol is used to solubilise the active substance tacrolimus (water solubility < 0.1 mg/mL) up to 5 mg/mL in Prograf® injection concentrate. The product is administered intravenously after dilution to concentrations of 0.004–0.1 mg/mL.
13.5.4 pH and Buffer Capacity
Solubility and stability of the active substances are to be considered when evaluating the optimal pH of parenteral solutions. Moreover the pH of the solution has an impact on the infusion tolerance, which is also depending on the volume and rate of administration and the buffer capacity of the formulation. To prevent possible adverse effects the pH should preferably be in the physiological range. The pH of the blood varies between 7.35 and 7.45 and has a large buffer capacity and, theoretically, a deviant pH of an intravenous injection will be corrected fast. The mixing, and thus the neutralisation process, however takes rather long as blood flows are laminar. Fast infusion will disturb this laminar flow and with irritating solutions might be preferred above slow infusion. This effect is confirmed in animal tests [20]. Intravenous injections have a higher chance of causing phlebitis in smaller vessels than in larger vessels where the blood flows faster. If the injection remains for too long at the place of injection such as with subcutaneous and intramuscular administration there is more chance of irritation than with intravenous administration.
If a larger volume is given intravenously, the tolerated pH range is 3–11 [1]. The more the pH of the formulation approximates to the lower and upper limit of this pH range the higher is the probability of discomfort and irritation at the injection site. Non-toxic acids and bases are used to set the pH of parenterals. Lactic, maleic and hydrochloric acid and sodium hydroxide are used most often. They have some buffering capacity in the acid area. Whether the resulting solution has sufficient buffering capacity depends on the pKa of the used base or acid and the pH of the solution (see Sect. 18.1.1).
Buffers, mostly phosphate buffer solution, are included in the formulations to ensure the pH of the solution being maintained throughout the shelf life of the product. The buffer prevents pH changes caused by degradation products or leaching of ions from glass containers (see Sect. 24.2.1). Buffers may increase the hydrolysis rate of the active substances (see Sect. 22.2.1) and may decrease solubility (Sect. 18.1.1). High buffer capacity should be avoided in order to minimise irritation and pain at the injection site and disturbance of the physiological pH.
However, due to active substance stability, it is still necessary to develop injections with extreme pH values. A currently licensed intravenous product in UK with a very low pH of 2.8 is Persantin® (dipyridamol 5 mg/mL). The solubility of dipyridamol is < 0.1 mg/mL, but higher at a pH ≤ 3.3 [21]. Experiments showed that even after dilution with 0.9 % NaCl or glucose 5 % solution (1:2 ratio), the pH still amounts to pH 3. Information about the pH of the diluted parenterals is not to be found in the literature for all licensed products [20].
13.5.5 Osmotic Value
As a measure of the tonicity of blood one can calculate with the osmotic value because active substances and additives cannot pass the membrane of the erythrocyte (see Sect. 18.5.2). The osmotic value of blood is around 290 mOsm/kg. Some parenteral fluids however contain substances that can pass the membrane fast: ethanol, glycerol, urea. Hyperosmotic solutions of these substances may cause haemolysis; so they are hypotonic. The iso-osmotic concentration of ethanol is for example 1.39 % m/m. Ethanol 5 % v/v infusion fluid is therefore hyperosmotic but appears to be practically isotonic.
Parenteral products with osmotic values differing from the physiological value may cause phlebitis and irritation. This is especially applicable when the injection remains relatively long at the site of injection such as after subcutaneous, intramuscular, epidural and intrathecal administration. There is a higher chance of phlebitis in small vessels after intravenous administration. However, it is not known which limits should be considered to prevent phlebitis and irritation. According to some sources [22] the osmolarity of an intravenous injection should not be higher than 500 mOsm/kg. For subcutaneous and intramuscular administration the range is smaller.
As is the case for the pH level and buffer capacity, adjustment of osmotic value to physiological conditions is advisable. Sodium chloride or glucose are mostly used to adjust tonicity of parenterals. Sometimes deviating medical requirements are to be met. For example, hyperbaric solutions of local anaesthetics are used in spinal anaesthesia (see Sect. 13.2.4). Hyperbaricity in comparison to the cerebrospinal fluid is achieved by a high glucose concentration (7.5 %) which is also a hyperosmolar.
Baricity and dose of the local anaesthetic along with the patient position determine the anaesthetic block height.
Nitroglycerin, Concentrate for Infusion 1 mg/ml
The alcoholic nitroglycerin solution 10 mg/g contains 90 % v/v ethanol (= 86.5 % m/m). The nitroglycerin infusion concentrate thus contains 86.5 mg ethanol per ml. It has an osmotic value of 1,800 mosmol/l and is thus hyper osmotic, but at the same time hypotonic because ethanol can pass the erythrocyte membrane freely. This solution can only be administered when it is mixed with NaCl 0,9 % or glucose 5 %.
13.5.6 Viscosity
Parenteral formulations should not be more viscous than water because as the viscosity of the formulation increases, the ease of administration decreases and the likelihood of pain caused by injection increases. Polymers that modify viscosity of the formulation are more frequently used in parenteral suspensions than in parenteral solutions. By addition of hydrophilic or lipophilic polymers parenteral suspensions are stabilised.
13.5.7 Excipients for Suspensions and Emulsions
Typical excipients used in parenteral suspensions include surfactants that are used to stabilise emulsions and suspensions as wetting agents (polysorbate 80, poloxamer), as micelle makers for the preparation of solubilisations and to influence the flocculation and deflocculation behaviour of a dispersed system (carmellose sodium, polyvidone). Parenterally used surfactants in high concentrations are toxic and may cause venous irritation and occasional thrombophlebitis. However, these high concentrations are not necessary to formulate stable parenteral suspensions.
Only a limited number of emulsifiers are commonly regarded as safe to use in emulsions for parenteral administration. Most important are lecithin, poloxamer and macrogolglycerol ricinoleate (Cremophor EL®). This latter excipient is linked to anaphylactic reactions and temporary haematological disorders.
13.5.8 Antioxidants
Antioxidants are used to reduce or inhibit the oxidation of active substances or excipients. Active substances in parenterals which may undergo oxidative degradation are e.g. phenothiazines, catecholamines, polyene antifungal agents, steroids, morphine. Antioxidants either prevent the formation of free radicals (i.e. butylated hydroxyanisole, butylated hydroxytoluene) or have a lower redox potential than the active substance to be protected (i.e. sodium metabisulfite, sodium formaldehydesulfoxylate). Edetate increases the activity of antioxidants by chelating polyvalent cations, which may generate free radicals or be involved in electron transfer reactions (see also Sects. 23.10 and 22.2.2.2). Another strategy to diminish oxidation is to remove oxygen by flushing the injection solution with nitrogen prior to closure (see Sect. 13.7.4).
Physostigmine salicylate can degrade successively by hydrolysis and oxidation by oxygen. To prevent oxidation during preparation, storage and administration to the patient physostigmine salicylate injection fluid (see Table 13.2) is stabilised with 0.1 mg/mL sodium metabisulfite. In general, to prevent oxidation sodium metabisulfite is used in higher concentrations i.e. 1 mg/mL. Because of the potential allergic reactions to sodium metabisulfite and the ability of metabisulfite to form adducts with physostigmine, the concentration is limited and arbitrarily set at 0.1 mg/mL. This concentration is adequate to protect the solution from discolouration.
Physostigmine salicylate | 0.1 g |
Hydrochloric acid | q.s. |
Sodium metabisulfite | 0.01 g |
Water for injections | ad 100 mL |
13.5.9 Preservatives
Single dose vials should be used as much as possible to avoid microbial contamination entering the vial as in the case of multidose vials. However, for economic reasons parenteral solutions are still packaged in multidose vials and preservatives are incorporated in them. According to the Ph.Eur. multidose vials must contain a preservative. However, the single dose may not exceed 15 mL. Parenteral solutions which contain high concentrations of co-solvents such as propylene glycol or glycerol, or oil-based products, mostly do not require the addition of preservatives, although it has been established that micro-organisms may survive in non-aqueous solutions [24]. The activity of preservatives can be affected by the pH, the concentration, the presence of binding or complexing excipients, and incompatibility reactions with proteins and rubber as container closure material. Commonly used preservatives and typical concentrations used are: benzyl alcohol (1.0 %), chlorobutanol (0.5 %), phenol (0.5 %), and metacresol (0.1 %). Some preservatives, such as the parabens, cause irritation or adverse reactions and are hardly used anymore in parenterals. If a formulation is intended for paediatric administration, restrictions for certain preservatives must be considered. European Medicines Agency (EMA) recommended that parenteral solutions containing benzyl alcohol should not be used in pre-term and full-term neonates, due to neurotoxicity, cardiovascular failure, intraventricular haemorrhage, gasping respirations and metabolic acidosis (‘gasping syndrome’). Due to the risk of fatal toxic reactions arising from exposure to benzyl alcohol in excess of 90 mg/kg/day, this preservative should also not be used in children up to 3 years old [25].
Due to their toxicity preservatives are not allowed in injections for epidural, intrathecal, intracisternal or intraocular use [26].
13.5.10 Excipients Used in Freeze-Drying
Proteinaceous active substances are usually formulated in an elaborated excipient system prior to the freeze-drying procedure. Bulking agents, such as mannitol or glycine, are used to increase the bulk volume and provide a suitable matrix in which a small quantity of protein is dispersed [27]. Further details regarding use of cryoprotective excipients are described in Sect. 22.2.5.
13.5.11 Packaging and Labelling
Injection fluids are packaged in ampoules, vials or bottles (container size ≤ 100 mL), syringes, and cartridges manufactured from glass or plastic. The containers must be transparent to allow visual inspection of the content. Light sensitive active substances should be preferably protected by light-tight secondary packaging; amber glass is an option as well but it hinders checking for clarity.
Containers for injections are discussed in Sects. 24.2.1 (glass quality), 24.2.4 (rubber), 24.3 (closures), 24.4.14 (ampoules) and 24.4.17 (syringes). Although glass is a preferred material, care should be taken to avoid possible incompatibilities. In particular, for instance, adsorption and aggregation of proteins touching glass surfaces, release of glass particles in solutions with pH > 7 and specific ions or both, release of aluminium from class I glass, etcetera. Class I glass is generally to be preferred but its suitability is not a matter of course and has to be tested. Rubber closures need attention as well because of release of rubber particles during autoclaving,
Because of the high price, type II glass is to be considered, and when tested might be found to be appropriate. Labelling of parenterals is discussed in Sect. 37.3.2. For injections usually both the total amount of active substance and the concentration should be indicated.
13.5.12 Stability
Details of chemical and physical degradation reactions of medicines (including proteins), and stability investigation are given in Chap. 22.
Injection solutions containing ‘small’ molecules may degrade by hydrolysis, oxidation and epimerisation reactions. The reaction rate determines whether sterilisation by heat can take place or not. Injection fluids that tolerate autoclaving usually have a shelf life of several years at room temperature. Active substances that are not stable in solution are usually prepared as powder – commonly by freeze drying the solution – and have to be reconstituted immediately before administration by the caregiver or the patient. Physical instability regards mainly suspensions (resuspendability) and protein solutions (precipitation and aggregation). Incompatibilities (precipitation) have to be checked for when solutions of poor water-soluble substances in co-solvents are diluted or when parenteral solutions are combined.
Protein products are especially prone to changes of the tertiary and quaternary structure of the molecules and thereby loss of activity. Degradation is induced by heat and shear stress. Rigorous shaking and administration by peristaltic pumps should be avoided.
13.5.13 Storage Temperature and Shelf Life
See Sect. 22.6.1 for a general discussion of shelf life. As long as the container of injection fluids remains closed, the storage temperature and shelf life are determined by the rate of the degradation reaction. Most parenterals can be stored at room temperature, exceptions are vaccines and protein medicines. Parenterals that are not sterilised in the final container are to be stored in the fridge or freezer with regard to maintaining their sterility. Storage temperature and in-use stability after first opening of ampoules and bottles are also determined by the chance of microbiological contamination.
The EMA Note for Guidance on Unpreserved Sterile Products requires licensed parenteral medicines to be labelled as follows:
Chemical and physical in use stability has been demonstrated for x hours/days at y °C. From a microbiological point of view, the product should be used immediately. If not used immediately, in-use storage times and conditions prior to use are the responsibility of the user and would normally not be longer than 24 h at 2–8 °C, unless reconstitution/dilution (etc.) has taken place in controlled and validated aseptic conditions.
The shelf life of reconstituted parenterals prepared in the hospital pharmacy is to be determined by the responsible pharmacist (see Sect. 22.6). When licensed injection fluids are reconstituted and prepared for individual patients the shelf life is determined by evaluation of the physico-chemical stability and risk of microbiological contamination. Shelf life depends from a microbiological viewpoint on the risks associated with aseptic preparation and the results of validation studies performed under the pharmacy’s specified aseptic preparation conditions (see Sect. 31.3.6).
In-use stability of preserved injection fluids in multiple dose vials is limited to a maximum of 28 days. Typical examples are multidose vials of insulin preserved with methyl hydroxyl benzoate and multidose vials of low molecular heparin preserved with benzyl alcohol.
13.5.14 Quality Requirements
Parenteral solutions are to be tested for absence of visible and non-visible particles and sterility as specified in the Ph. Eur. Furthermore, freedom from pyrogens or endotoxins and the uniformity of dosage units and content are stipulated (see Sect. 32.8). Suspensions should be easily resuspendable to ensure precise dosing and uniformity of the dose during administration. Parenteral emulsions should be homogeneous.
13.5.15 Special Parenteral Preparations
In practice the pharmacist may dispense special injections. This section briefly describes their formulation and biopharmaceutic characteristics, but not their way of preparation and quality requirements.
13.5.15.1 Suspensions
Parenteral suspensions are injected subcutaneously or intramuscularly and are usually meant for an extended action [28]. Examples of parenteral suspensions licensed in Europe are penicillin G procaine suspension and medroxyprogesterone acetate suspension.
13.5.15.2 Gels
Biodegradable in situ gel forming implant systems secure prolonged activity. They are formulated by dissolving biodegradable polymers (such as poly (dl-lactide), lactide/glycolide copolymers, lactide/caprolactone copolymers) in a biocompatible organic solvent (e.g. N-methyl-2-pyrrolidone (NMP)). Active substances are either dissolved or dispersed in the polylactic-co-glycolic acid (PLGA) solution. When the liquid formulation is injected intramuscularly or subcutaneously, the organic solvent diffuses into body fluid and water penetrates into the solution. This leads to precipitation of the polymer forming a solid polymeric implant at the injection site. The active substance is released over a prolonged period when the polymer is biodegraded [29].
An advanced injectable gel product is Eligard® containing leuprolide acetate and PLGA 75/25 dissolved in NMP in a 45:55 (w/w) ratio. The 1, 3 and 4 months Eligard® formulations are supplied in two separate syringes. One syringe contains the PLGA/NMP solution and the second syringe leuprolide acetate. Prior to administration the content of two syringes is mixed until it is homogeneous.
13.5.15.3 Implants
Implants are solid polymers loaded with an active substance. The active substance is delivered at a constant rate as long as the implant stays at the administration site. Local or systemic activity can be achieved. The duration of activity varies from days to years. Insertion and removal (if not biodegradable) requires medical assistance [30].
In order to achieve high local concentrations of antibiotics in infected bone or joint, methyl methacrylate/methyl acrylate copolymer (PMMA) bead chains or mini-chains loaded with an antibiotic (mostly gentamicin sulfate) are inserted.
Gentamicin sulfate is heat resistant and highly soluble in water. A bead chain consists of 10, 30 or 60 beads (diameter 7 mm) threaded on a flexible steel thread. A mini-chain contains 10 or 20 oval mini beads threaded over a length of 10 or 20 cm. The beads are sterilised with ethylene oxide. The chain should be removed after a period of 7–10 days.
Another typical implantable product is the horse collagen sponge loaded with gentamicin. The implant is absorbable. High antibiotic concentrations are achieved at the site of implantation over several days.
13.5.15.4 Derivatives
Protein and peptide pegylation (conjugation to polymers of long-chain macrogol – or polyethylene glycol) improves the biopharmaceutical properties of active substances. The improvement encompasses increased stability, resistance to proteolytic inactivation and extension of the half-lives. By this the delivery and efficacy of proteins is improved [31].
The application of filgrastim pegylation technology resulted in a second generation molecule named pegfilgrastim, with significantly altered pharmacokinetic properties in comparison to filgrastim. Pegfilgrastim has the same mechanism of action as filgrastrim but the pegylation markedly reduces renal clearance, leaving neutrophil-mediated clearance as the major route of elimination. As a result, clearance of pegfilgrastim is decreased and serum concentrations are sustained throughout the duration of neutropenia. In clinical studies with standard, multicycle chemotherapy, a single subcutaneous dose of pegfilgrastim has been shown to have similar efficacy and safety as daily subcutaneous injections doses of filgrastim in patients with solid tumours [32].
Further examples of pegylated protein active substances are peginterferon and pegvisomant.
Polysialylation (the process whereby PSA-polysialic acid is conjugated with proteins) is an alternative to pegylation limiting potential toxic accumulation and immunogenic potential of macrogol [33].
13.6 Product Formulation of Infusions
Many formulation characteristics and quality requirements for infusions are the same as for injections.
13.6.1 Types of Infusions
Infusions are sterile solutions or emulsions that are administered intravenously, subcutaneously or intrathecally/epidurally. Infusions are used as:
Solutions regulating the acid–base balance
Vehicle solutions for continuous infusion
Blood volume expanders
Osmotic diuretics
Partial and total parenteral nutrition
Shifts of the physiological pH may require administration of acid or base containing infusions. Metabolic acidosis is treated by infusion of sodium bicarbonate containing infusion solutions. Metabolic alkalosis is treated with hydrochloric acid containing infusion solutions.
Sodium chloride 0.9 % solution and 5 % glucose solutions are most suitable to be used as vehicle solutions for parenterally administered injections or infusions.
Blood volume expanders can be crystalloid or colloidal. The most often used crystalloid solutions are 0.9 % sodium chloride solution, 5 % glucose solutions and their combinations, e.g. 0.45 % sodium chloride/2.5 % glucose solution. In addition infusion solutions containing multiple electrolytes such as Ringer’s solution or Lactated Ringer’s are administered. These solutions contain also potassium, magnesium, calcium, and phosphate [34].
Blood volume expanders used to compensate loss of plasma are formulated as electrolyte solutions with macromolecular substances such as gelatine, polygeline, hydroxyethyl starch (HES), and albumin. Hydroxyethyl starch is similar to albumin but cheaper. However there are restrictions in its use. European Medicine Agency (EMA) has decided that HES solutions should not be used in critically ill adult patients, including patients with sepsis or burn injuries and excess bleeding particularly in patients undergoing open heart surgery in association with cardiopulmonary bypass, because an increased risk of mortality and renal injury requiring renal replacement therapy [35].
Osmotic diuretics are rapidly filtered in the glomerulus and not reabsorbed in the tubules. As a result of the osmotic gradients thereby achieved in the renal tubules, the reabsorption of water is inhibited, and urine excretion increased (see also Sect. 13.6.3).
Parenteral nutrition solutions are discussed separately in Sect. 13.9.
13.6.2 Buffer Capacity
Large volume infusions should have a physiological pH, e.g. pH 7.4 and little or no buffer capacity. In general infusions with pH 5–9 are tolerated by peripheral veins.
The pH values of commonly used infusion solutions vary between 4 and 8 depending on the type of formulation. A lower pH is acceptable if solutions are not buffered. The pH of 5 % glucose solutions amounts from 4.0 to 5.0. This is necessary to prevent degradation of glucose (see Sect. 13.6.4). Infusion fluids that are used to correct the pH of the blood have a deviant pH value (e.g. pH 8.0 for sodium hydrogen carbonate infusion).
13.6.3 Osmolarity
See Sect. 18.5 about osmosis and tonicity.
Infusion solutions have to be isotonic with blood because of the large volume administered. Typically used isotonic infusion solutions are 0.9 % NaCl solution and 5 % glucose solution.
Hyperosmotic mannitol 20 % infusion is used as osmotic diuretic. Hypotonic solutions such as 0.65 % sodium chloride are used in elderly patients to keep the salt load low, or in children to prevent dehydration.
Infusion fluids for neonates are often hyperosmotic, having no choice since only small volumes can be administered.
According to some recommendations [36] if osmolarity of the infusion is higher than 500 mOsmol/l, administration should occur via a central venous catheter that is inserted in a vessel with high blood flow such as the vena subclavia, proximal axillary vein or superior vena cava.
13.6.4 Stability
In addition to what has been said about the stability of injections (Sect. 13.5.12) some specific comments can be given regarding infusions. In practice most concern is about the chemical and physical (precipitation!) stability of active substances when mixing injections with the infusion vehicle. As a consequence of incompatibilities some active substances are only compatible with 0.9 % NaCl vehicle (e.g. furosemide-Na injection concentrate) and others are only compatible with 5 % glucose solution. As the mixtures often have to be kept after mixing also the chemical stability is an issue as well as minimising the chance of microbiological contamination by proper aseptic handling and administration.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


