Chapter 6 Diffuse parenchymal disease of the lung
Diffuse parenchymal lung disease in childhood
Some diseases that affect the periphery of the lung largely involve the alveolar interstitium whereas others encroach upon, or preponderate within, the alveolar lumen. Until the advent of modern imaging techniques this distinction was not fully appreciated by clinicians, with the result that many diseases have been termed interstitial, irrespective of the lung compartment predominantly involved. A classification formulated by a joint committee of the American Thoracic Society and the European Respiratory Society in 2002 (the ATS/ERS classification) corrects this anomaly by referring to them as the diffuse parenchymal lung diseases.1
The parenchyma of an organ is the part concerned with function. In most viscera, function resides in the epithelium, and the interstitial connective tissue merely acts in a supporting role. This is not the case in the lungs. The function of the lungs is of course gas exchange, which takes place in the alveoli. It is unlikely that gas exchange could take place if any component of the alveolus or its wall was lacking. The lung parenchyma is therefore to be regarded as all the tissue of the gas-exchanging region, which is that portion beyond the terminal bronchiole – the pulmonary acinus (see p. 4 and Fig. 1.4, p. 5). An earlier term synonymous with diffuse parenchymal lung disease is acinar lung disease.2 The lung acinus includes the alveolar interstitium and endothelium as well as its epithelium and air space. Thus the term ‘diffuse parenchymal (or acinar) lung disease’ is highly appropriate for a group of diseases that includes some which are truly interstitial and others that represent alveolar filling defects.
The ATS/ERS classification concentrates upon the idiopathic interstitial pneumonias and in this area seeks to match disease diagnosis with predominant histological pattern so that there is often dual terminology (Table 6.1).1 It brings together clinical, imaging and histological criteria and is of undoubted prognostic and therapeutic value. However, it is not above criticism, particularly in its retention of terms such as ‘interstitial pneumonia’ for a group of parenchymal diseases that may be either interstitial or intra-alveolar. The term ‘desquamative interstitial pneumonia’ (DIP) is retained, although it is now well established that this process is not desquamative but exudative and has only a minor interstitial component. Cryptogenic organising pneumonia is a further condition listed under the idiopathic interstitial pneumonias despite it affecting the alveolar space rather than the interstitium. Furthermore, DIP and respiratory bronchiolitis are largely caused by smoking and are therefore not strictly idiopathic. Similarly, lymphoid interstitial pneumonia (LIP) is virtually never idiopathic and it is questionable whether it should be regarded as an interstitial pneumonia or a lymphoproliferative disease.
Table 6.1 Classification of diffuse parenchymal lung disease1
1 Of known cause, e.g. drugs (Chapter 7.3) or of known association, e.g. collagen vascular disease (Chapter 10) |
| Diagnosis | Predominant histological pattern |
|---|---|
| Idiopathic pulmonary fibrosis (cryptogenic fibrosing alveolitis) | Usual interstitial pneumonia |
| Non-specific interstitial pneumonia (provisional)a | Non-specific interstitial pneumonia |
| Desquamative interstitial pneumonia | Desquamative interstitial pneumonia |
| Respiratory bronchiolitis-associated interstitial lung disease | Respiratory bronchiolitis |
| Acute interstitial pneumonia | Diffuse alveolar damage |
| Cryptogenic organising pneumoniab | Organising pneumonia |
| Lymphoid interstitial pneumonia | Lymphoid interstitial pneumonia |
a A heterogeneous group of diseases with poorly characterised clinical and radiological features that need further study.
b Synonymous with idiopathic bronchiolitis obliterans organising pneumonia.
The prime reason for assembling the seven disparate entities listed in Table 6.1 under the heading idiopathic interstitial pneumonias is that, although they differ in their prognosis and treatment, they may enter the differential diagnosis of idiopathic pulmonary fibrosis (IPF) at initial presentation. These patterns are also of considerable prognostic value.1,3–8 The ATS/ERS classification provides pathological criteria that allow these patterns to be distinguished in a fairly reproducible manner.9 However, pathologists should be aware that they are only identifying a particular histological pattern, not a specific disease, subsequent correlation with clinical and imaging data being required to reach a final diagnosis.1 Mixed patterns may be encountered. For example, diffuse alveolar damage, the pathological basis of acute interstitial pneumonia (AIP), may be superimposed on UIP in acute exacerbations of IPF,10 different lobes may concurrently show different histological patterns in the same patient11 and different histological patterns may be seen in successive samples of the same lung.12 However, there is nothing to stop pathologists reviewing all the data and coming to a final diagnosis themselves.13
It is also worth noting that, whereas a UIP pattern is typical of IPF, conditions other than IPF may show UIP on occasion, while diffuse alveolar damage has many recognisable causes besides being the basis of AIP (see Box 4.2, p. 143) and NSIP is seen in a variety of unrelated conditions.3 One review estimated that 68% of cases showing NSIP were idiopathic, 22% were associated with connective tissue disease, 8% represented atypical cases of extrinsic allergic alveolitis and 2% followed the acute respiratory distress syndrome.14 Another group estimated that 88% of patients classified as idiopathic NSIP had what they termed unclassified connective tissue disease.15
The terms ‘acute interstitial pneumonia’ (AIP) and NSIP are relatively new and when the adjective ‘usual’ was originally introduced it was to contrast UIP with four other patterns of idiopathic interstitial pneumonia, namely DIP, LIP, a pattern occurring in association with bronchiolitis obliterans (BIP) and one marked by the presence of giant cells (GIP).16 It was hoped that this histopathological classification would facilitate a better understanding of aetiology, pathogenesis and clinical features. This hope has been partly realised in that many cases of GIP are now known to be caused by hard-metal alloys. The term BIP is no longer used, having been superseded initially by idiopathic bronchiolitis obliterans organising pneumonia and now by cryptogenic organising pneumonia.
A further pattern, described since the ATS/ERS classification was formulated, is one that has been termed bronchiolocentric interstitial pneumonia.17–20 This lacks any specific features and is of unknown cause but before using the term bronchiolocentric interstitial pneumonia, the pathologist should consider the possibility that the changes may represent inactive forms of such centriacinar diseases as extrinsic allergic alveolitis. Having done so, one group suggested that the changes were the result of gastric aspiration.18
The term cryptogenic fibrosing alveolitis (CFA) was introduced in the 1960s to link IPF to the pulmonary fibrosis that is occasionally seen in association with the collagen vascular disorders, the prefixes ‘lone’ and ‘system’ being used to distinguish the two.21–23 However, the prefixes are now seldom used and the unqualified term CFA has become an unnecessary synonym for IPF1 or is used as a general umbrella term for pulmonary fibrosis of uncertain aetiology.24
Antedating all these terms but now largely abandoned are those applied by nineteenth-century morbid anatomists who were evidently familiar with the pathological changes seen in IPF and applied graphic descriptive terms such as muscular cirrhosis of the lung25 and cirrhosis cystica pulmonum.26
Diffuse parenchymal lung disease in children is very different from that seen in adults. IPF with its characteristic pattern of UIP is virtually unknown in children,27,28 most reports antedating the ATS/ERS consensus classification. When the criteria recommended in this classification are applied to the paediatric cases it is found that patterns other than UIP are more common. AIP is also very rare in children, although its histological pattern of diffuse alveolar damage was first recognised in premature babies dying of the infantile respiratory distress syndrome. On the other hand, LIP is common in childhood, where it is nearly always associated with either connective tissue disease or immunodeficiency, both congenital and acquired.27,28 It is a well-described complication of paediatric acquired immunodeficiency syndrome (AIDS), occurring in over 30% of children infected perinatally by human immunodeficiency virus (HIV). NSIP is also increasingly recognised in children and some cases are a result of surfactant gene mutations. DIP is rare and its outcome is worse in children compared to adults. Some childhood cases of DIP represent inborn errors of metabolism such as surfactant B deficiency and lipid storage diseases.29–31 Cryptogenic organising pneumonia is exceptionally rare in children and respiratory bronchiolitis has not been reported. Disorders such as extrinsic allergic alveolitis and eosinophilic pneumonia are uncommon in children but have to be considered in the differential diagnosis, as do infections and their sequelae, which tend to be diffuse in this age group. Langerhans cell histiocytosis is seen in children with the systemic forms of the disease, especially Letterer–Siwe disease, but the type confined to the lung is largely a disease of cigarette smokers. After all these conditions have been considered there remain diffuse parenchymal diseases that feature in the perinatal period: these are considered in Chapter 2 (see Table 2.2, p. 51).32,33
1 American Thoracic Society/European Respiratory. Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2002;165:277-304.
2 Scadding JG. Talking clearly about diseases of the pulmonary acini. Br J Dis Chest. 1978;72:1-12.
3 Katzenstein ALA, Fiorelli RF. Nonspecific interstitial pneumonia/fibrosis – histologic features and clinical significance. Am J Surg Pathol. 1994;18:136-147.
4 Bjoraker JA, Ryu JH, Edwin MK, et al. Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998;157:199-203.
5 Katzenstein ALA, Myers JL. Idiopathic pulmonary fibrosis: Clinical relevance of pathologic classification. Am J Respir Crit Care Med. 1998;157:1301-1315.
6 Nicholson AC, Colby TV, Dubois RM, et al. The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med. 2000;162:2213-2217.
7 King TE, Schwarz MI, Brown K, et al. Idiopathic pulmonary fibrosis – Relationship between histopathologic features and mortality. Am J Respir Crit Care Med. 2001;164:1025-1032.
8 Flaherty KR, Toews GB, Travis WD, et al. Clinical significance of histological classification of idiopathic interstitial pneumonia. Eur Resp J. 2002;19:275-283.
9 Nicholson AG, Addis BJ, Bharucha H, et al. Inter-observer variation between pathologists in diffuse parenchymal lung disease. Thorax. 2004;59:500-505.
10 Kondoh Y, Taniguchi H, Kawabata Y, et al. Acute exacerbation in idiopathic pulmonary fibrosis – analysis of clinical and pathologic findings in three cases. Chest. 1993;103:1808-1812.
11 Flaherty KR, Travis WD, Colby TV, et al. Histopathologic variability in usual and nonspecific interstitial pneumonias. Am J Respir Crit Care Med. 2001;164:1722-1727.
12 Katzenstein ALA, Zisman DA, Litzky LA, et al. Usual interstitial pneumonia – Histologic study of biopsy and explant specimens. Am J Surg Pathol. 2002;26:1567-1577.
13 Myers JL, Katzenstein ALA. Beyond a consensus classification for idiopathic interstitial pneumonias: progress and controversies. Histopathology. 2009;54:90-103.
14 Cottin V, Loire R, Chalabreyesse L, et al. Pneumopathie interstitielle non specifique: une nouvelle entité anatomoclinique au sein des pneumopathies interstitielles diffuses idiopathiques. Rev Mal Respir. 2001;18:25-33.
15 Kinder BW, Collard HR, Koth L, et al. Idiopathic nonspecific interstitial pneumonia: lung manifestation of undifferentiated connective tissue disease? Am J Respir Crit Care Med. 2007;176:691-697.
16 Liebow AA. New concepts and entities in pulmonary disease. International Academy of Pathology monograph No. 8. The Lung. Williams & Wilkins, Baltimore. 1967;332-365.
17 Yousem SA, Dacic S. Idiopathic bronchiolocentric interstitial pneumonia. Modern Pathol. 2002;15:1148-1153.
18 deCarvalho MEP, Kairalla RA, Capelozzi VL, et al. Centrilobular fibrosis: A novel histological pattern of idiopathic interstitial pneumonia. Pathol Res Pract. 2002;198:577-583.
19 Churg A, Myers J, Suarez T, et al. Airway-centered interstitial fibrosis – A distinct form of aggressive diffuse lung disease. Am J Surg Pathol. 2004;28:62-68.
20 Fukuoka J, Franks TJ, Colby TV, et al. Peribronchiolar metaplasia: a common histologic lesion in diffuse lung disease and a rare cause of interstitial lung disease: clinicopathologic features of 15 cases. Am J Surg Pathol. 2005;29:948-954.
21 Scadding JG. Fibrosing alveolitis. BMJ. 1964;2:686.
22 Scadding JG, Hinson KFW. Diffuse fibrosing alveolitis (diffuse interstitial fibrosis of the lungs): correlation of histology at biopsy with prognosis. Thorax. 1967;22:291-304.
23 Scadding JG. Diffuse pulmonary alveolar fibrosis. Thorax. 1974;29:271-281.
24 Wells AU, Hansell DM, Nicholson AG. What is this thing called CFA? Thorax. 2007;62:3-4.
25 Buhl VL. Muscular Zirrhose der Lungen. Lungenentzundung, Tuberkulose und Schwindsucht. Munich: R. Olderburg; 1873. p. 169
26 Rindfleisch GE. Ueber Cirrhosis Cystica Pulmonum. Zentralbl Pathol. 1897;8:864-865.
Diffuse parenchymal lung disease in childhood
27 Fan LL, Kozinetz CA, Wojtczak HA, et al. Diagnostic value of transbronchial, thoracoscopic, and open lung biopsy in immunocompetent children with chronic interstitial lung disease. J Pediatr. 1997;131:565-569.
28 Nicholson AG, Kim H, Corrin B, et al. The value of classifying interstitial pneumonitis in childhood according to defined histological patterns. Histopathology. 1998;33:203-211.
29 Amir G, Ron N. Pulmonary pathology in Gaucher’s disease. Hum Pathol. 1999;30:666-670.
30 Stillwell PC, Norris DG, O’Connell EJ, et al. Desquamative interstitial pneumonitis in children. Chest. 1980;77:165-171.
31 Tal A, Maor E, Bar-Ziv J, et al. Fatal desquamative interstitial pneumonia in three infants siblings. J Pediatr. 1984;104:873-876.
32 Deutsch GH, Young LR, Deterding RR, et al. Diffuse Lung Disease in Young Children: Application of a Novel Classification Scheme. Am J Respir Crit Care Med. 2007;176:1120-1128.
33 McFetridge L, McMorrow A, Morrison PJ, et al. Surfactant metabolism dysfunction and childhood interstitial lung disease (chILD). Ulster Med J. 2009;78:7-9.
6.1 Interstitial lung disease
The diseases described in this section predominantly involve the alveolar interstitium, which constitutes the connective tissue framework of the alveolar walls. Its boundaries are the epithelial and endothelial basement membranes (see Fig. 1.28, p. 17). Many of the diseases considered here involve the interstitial fibroblasts and progress to interstitial fibrosis and ultimately ‘honeycombing’ (see p. 149). They represent examples of restrictive lung disease, the other major cause of which is interstitial oedema, which is dealt with in Chapter 8.1 (p. 401). Lymphangioleiomyomatosis is an exception in that it progresses to severe cystic change without any appreciable degree of fibrosis.
Usual interstitial pneumonia and idiopathic pulmonary fibrosis
Usual interstitial pneumonia (UIP) is the commonest histological pattern seen in idiopathic interstitial pneumonia. It generally correlates clinically with idiopathic pulmonary fibrosis (IPF). However, it may also, on rare occasions, be encountered in drug reactions, collagen vascular diseases and hypersensitivity pneumonia, and IPF can only be diagnosed when these possibilities have been excluded.1–3 Some regard UIP as the only acceptable pattern in IPF,1 but others report fibrotic non-specific interstitial pneumonia (NSIP) in a minority of patients with this disease.4–7 Occasionally, UIP is observed in one lobe and NSIP in another, in which case the prognosis is that of UIP.4,7 Although abundant macrophages may accompany UIP, desquamative interstitial pneumonia (DIP) is now considered to represent a separate process, which is dealt with on page 311.5,8–11
Epidemiology
IPF shows a wide age spectrum. The mean age at presentation is 71 years,12 somewhat older than earlier studies that possibly included patients with DIP.13,14 The disease affects twice as many men as women and there is a strong association with smoking.14–16 Prevalence is between 1 and 5 per 100 000 population.17 In the UK the reported incidence doubled between 1990 and 2003 while in the USA age-adjusted death rates over a similar period increased 28% in men and 41% in women.12,18 There is no predilection for specific countries, ethnic groups or urban/rural locations.1 Occasionally, several members of a family are affected.19,20
Clinical features
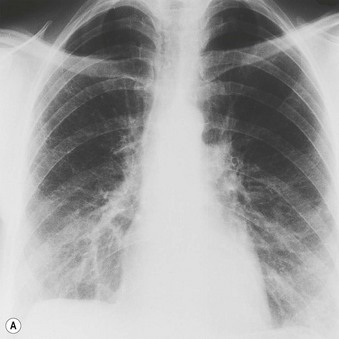
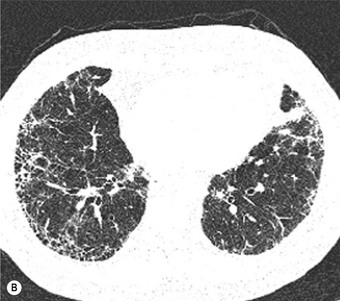
Most patients with IPF complain of breathlessness on exertion, dry cough and loss of weight.16 Bilateral basal crackles are heard on inspiration and digital clubbing is commonly found. Functional studies show a restrictive respiratory defect, the lungs being small and stiff. Chest X-rays may show a ‘ground-glass’ pattern or fine linear mottling, predominantly affecting the subpleural region of the lung bases. Progressive elevation of the diaphragm and ‘honeycombing’ accompany the shrinkage of the fibrotic lung tissue (Fig. 6.1.1A). On high-resolution computed tomography (HRCT), the dominant pattern comprises irregular lines forming a reticular pattern, mainly involving the bases of the lungs. As the disease progresses, the reticular pattern becomes coarser and ‘honeycombing’ and ‘traction bronchiectasis’ develop (Fig. 6.1.1B). Serological abnormalities and changes detectable on bronchoalveolar lavage (outlined below under pathogenesis) contribute to a distinctive clinical picture.
The disease is generally diagnosed on the above clinical and imaging features; indeed, the diagnostic accuracy of HRCT in identifying patients with a UIP pattern and hence IPF is now such that many patients do not have a biopsy.1,21 In the UK only 40% of patients with IPF undergo any form of biopsy: 28% have a transbronchial biopsy and 12% a surgical lung biopsy.16 Transbronchial biopsy may establish an alternative diagnosis, but a surgical biopsy is nearly always required to determine the pattern of interstitial pneumonia, and is now usually obtained at video-assisted thoracoscopy. In these instances, it is the presence of UIP that establishes the diagnosis (of IPF) and dictates prognosis.22
Course of the disease
Early studies showed a mean survival of about 4 years14 but in later series limited to patients with UIP this figure was reduced to about 2 years (Fig. 6.1.2).1,5,8,22,23 The later stages of the disease are characterised by cyanosis and dyspnoea at rest. These features presage death from respiratory failure, cor pulmonale or lung cancer, this last being an important complication that is dealt with below. Although IPF is typically characterised by a steady, predictable decline, some patients experience more rapid deterioration, either episodically or as a terminal event.3,24–32 These acute exacerbations are defined as clinically significant deteriorations in patients with underlying IPF, characterised by subjective worsening over 30 days or less, new bilateral radiographic opacities and the absence of infection or another identifiable aetiology.31
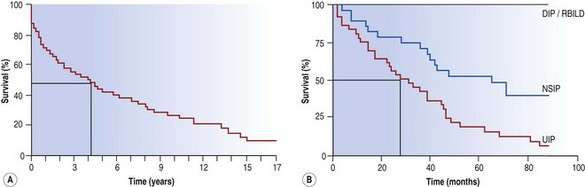
Figure 6.1.2 The survival curve of two groups of patients treated for idiopathic pulmonary fibrosis at the Brompton Hospital. (A) In a group antedating the distinction of usual interstitial pneumonia (UIP) and non-specific interstitial pneumonia (NSIP) the median survival was about 4 years from the onset of symptoms whereas in a later group (B) categorised according to their histological pattern, those with a UIP pattern had a median survival nearer to 2 years. DIP, desquamative interstitial pneumonia; RBILD, respiratory bronchiolitis-associated interstitial lung disease.
Until recently, treatment was largely based on immunosuppression but this has seldom proved effective23,33 and in the last decade several other drugs have either been or are being assessed.3 These include interferon-γ, which initially showed promise34 but has since shown no advantage.35 In recent trials neither etanercept (a tumour necrosis factor-α antagonist) nor imatinib (a tyrosine kinase inhibitor) influenced the primary study endpoint, although both showed some evidence of decreasing disease progression.36,37 More promising was a study with N-acetyl cysteine in combination with steroids and azathioprine that achieved its primary endpoint in showing a significant slowing of disease progression,38 and initial trials of both bosentan (an endothelin-1 receptor antagonist)39 and pirfenidone40,41 have suggested a positive effect. Another agent to show a positive treatment effect, notably a reduction in mortality, has been warfarin.42 The introduction of these drugs reflects advances in our understanding of the pathogenesis of IPF that have in part arisen from the more precise definition of the disease within the consensus classification.43–47 Currently however, the only treatments that can be recommended outside a drug trial are oxygen therapy and transplantation.3
Morbid anatomy
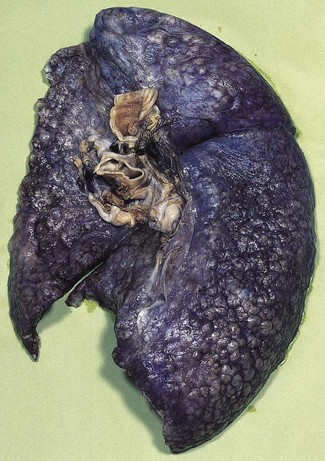
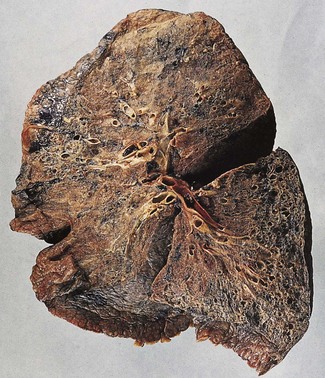
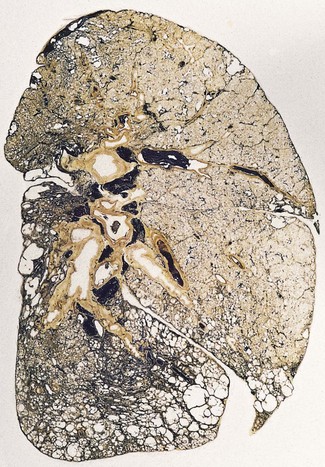
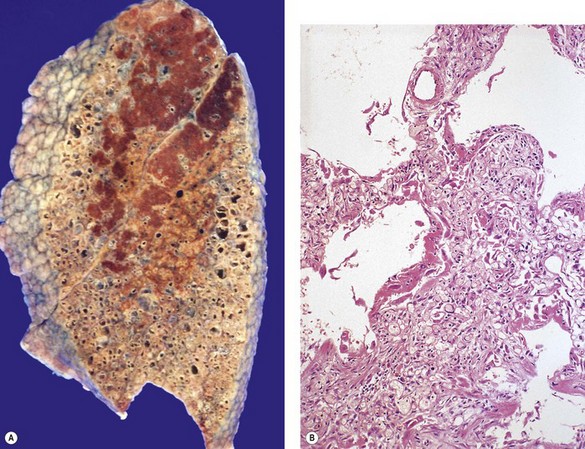
In a typical case, the lungs are shrunken and firm when removed at autopsy or in preparation for transplantation. The lower lobes are most severely affected, with the pleura having a finely nodular ‘cobblestone’ pattern, resembling that of a cirrhotic liver (Fig. 6.1.3). Pleural fibrosis is uncommon, in contrast to asbestosis which IPF otherwise resembles macroscopically. The cut surface of the lung shows fibrosis and a variable degree of ‘honeycombing’, which is most marked beneath the pleura (Figs 6.1.4 and 6.1.5). The posterior basal segment is maximally affected and from here the process extends upwards, involving particularly the subpleural lung tissue. This rind of contracted fibrous tissue prevents the lungs from expanding and is an important factor contributing to the restrictive respiratory defect.
Histological changes
The cardinal features of UIP2 are:
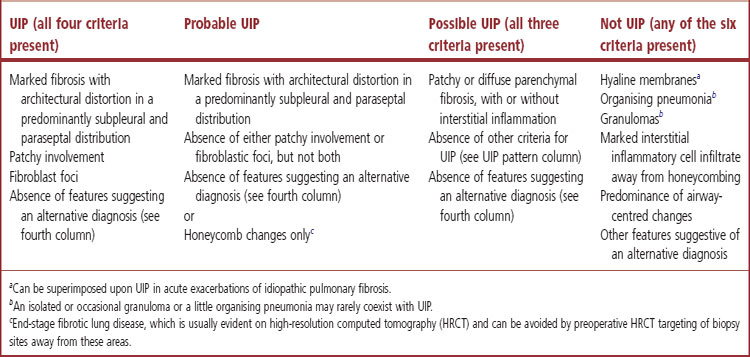
As noted above, the disease is most marked immediately beneath the pleura and characteristic low-power feature is that the changes are patchy. Areas of chronic interstitial inflammation and fibrosis alternate with foci of relatively normal lung (Fig. 6.1.6). Much of the fibrosis is well established, consisting of poorly cellular hyaline collagen, but lying immediately adjacent to this older fibrous tissue are localised areas of granulation tissue consisting of plump fibroblasts set in a slightly haematoxyphilic myxoid stroma (Fig. 6.1.7). This variation in the age of the fibrosis is sometimes described as evidence of ‘temporal heterogeneity’. The foci of immature fibrous tissue, termed ‘fibroblastic foci’, can be highlighted by staining the abundant acidic proteoglycans present in such granulation tissue with alcian blue or Movat’s stain (see Fig. 6.1.7C). They are thought to represent the sites of repeated lung damage.13 Their progression is characterised by weakening alcianophilia and increasing collagenisation.48 Most studies suggest that a profusion of these foci predicts poor survival whereas the more traditional feature of disease activity, interstitial mononuclear cell infiltration, is of only limited prognostic value.49–52 Despite the term ‘interstitial pneumonia’, chronic inflammatory cell infiltration is generally only mild to moderate in intensity. Lymphoid follicles are not usually prominent in idiopathic disease and when present suggest collagen vascular disease.53 As the process becomes more advanced, there is increased loss of alveolar architecture with remodelling and eventual formation of cysts separated by bands of fibrosis, so-called honeycombing.
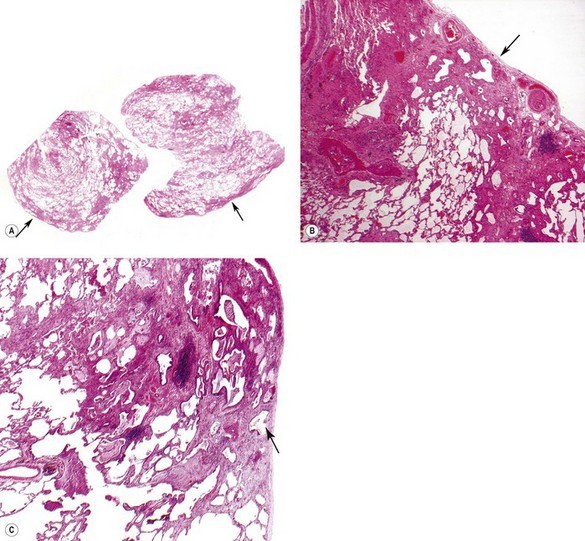
Figure 6.1.6 Usual interstitial pneumonia. Patchy subpleural and paraseptal fibrosis is characteristic of this pattern of interstitial pneumonia. In all cases, the fibrosis is interspersed with unaffected normal lung. Arrows pleura.
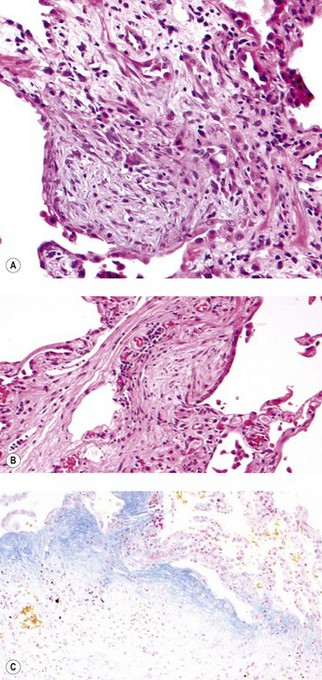
Figure 6.1.7 Usual interstitial pneumonia. (A) A fibroblastic focus consisting of granulation tissue adjacent to more collagenous interstitial fibrosis. (B) A fibroblastic focus showing a greater degree of collagenisation and overlying epithelialisation as it becomes incorporated in the interstitium. (C) Granulation tissue rich in alcianophilic proteoglycans (top) covers older, more mature fibrous tissue (Alcian blue stain).
This full constellation of histological features permits the pathologist to recognise a UIP pattern with confidence but in some cases only some of these features are evident, in which case only an opinion of probable or possible UIP can be offered. However, histological assessment is also of value in that it sometimes permits the confident exclusion of UIP (Table 6.1.1, p. 272).3
Although widespread ‘honeycombing’ usually represents advanced IPF, attempts to differentiate end-stage UIP from end-stage NSIP have shown poor reproducibility5 and it is preferable to classify these changes merely as end-stage lung rather than assign a particular histological pattern.5,54 Such areas can often be avoided if the surgeon discusses the cases with radiologist and pulmonologist before taking the biopsy to ascertain the probable sites of active disease.2 Many centres now biopsy two sites to reduce the possibility of sampling error.4,7
Patients with IPF frequently smoke and it is therefore not surprising that a variety of further changes commonly found in smokers may be seen in addition to UIP, namely centriacinar emphysema, respiratory bronchiolitis, DIP-like changes and what has been termed air space enlargement with fibrosis.56
Electron microscopy
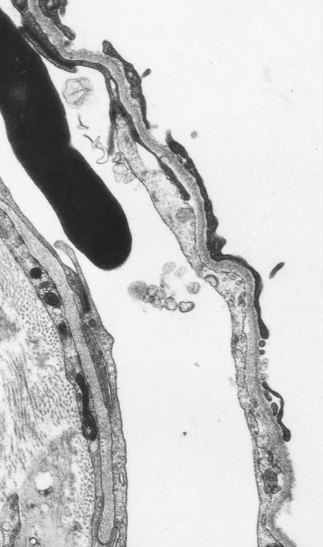
Ultrastructural studies support the view that the alveolar epithelium is the target tissue, identifying foci of bare epithelial basement membrane as an early feature of the disease (Fig. 6.1.9).57–59
Differential diagnosis
IPF was formerly categorised as either ‘cellular’ or ‘fibrotic’ and, although this was of prognostic value,14,60,61 the ‘cellular’ cases would now be recognised as having a different histological pattern, such as cellular NSIP, DIP or respiratory bronchiolitis. It is important to distinguish UIP from these other patterns of interstitial pneumonia because IPF differs radically from the other interstitial lung disease in its prognosis and treatment (see Fig. 6.1.2). The most problematic area is the distinction of UIP from fibrotic NSIP.62 The key features in this are a relatively diffuse pattern of interstitial fibrosis and an absence or dearth of fibroblastic foci (lack of temporal heterogeneity) in NSIP.2 In reality, this can be very difficult and diagnostic confidence is often low,62 not helped by the fact that, as well as areas showing UIP and normal lung, foci indistinguishable from NSIP are often seen.6 In these difficult cases, consideration of the clinical and imaging features may be of great help.62a
Sometimes there are abundant macrophages, resulting in a DIP-like pattern, but attention to the alveolar walls reveals the characteristic fibroblastic foci and patchy subpleural distribution of UIP.2,63,64 Any fibrosis found in DIP is mild, diffuse and non-fibroblastic.
Diffuse alveolar damage (DAD) is easily distinguished by its characteristic hyaline membranes but may be superimposed upon UIP in acute exacerbations of IPF (Fig. 6.1.10).25,27–31
UIP differs from asbestosis by the absence of asbestos bodies, from granulomatous diseases such as Langerhans cell histiocytosis and sarcoidosis by the absence of granulomas, and from lymphangioleiomyomatosis by the different nature of the smooth muscle, which is atypical and differs markedly from that seen as reactive hyperplasia in IPF (compare Figs 6.1.8C and 6.1.48).
Extrinsic allergic alveolitis can also usually be excluded by the presence of granulomas, but chronic cases may be difficult to distinguish from UIP.65,66 This again emphasises the need to correlate the biopsy findings with clinical and imaging data.67
If the vascular changes are so severe that primary pulmonary hypertension enters the differential diagnosis, attention should be paid to the vessels in the comparatively normal areas of the biopsy, when it will generally be appreciated that the abnormal blood vessels are limited to areas of diseased parenchyma, unlike the generalised vascular hypertrophy seen in primary pulmonary hypertension. Pulmonary hypertension can of course complicate IPF, when it is an adverse prognostic indicator.68
Occasionally, lung biopsy reveals fibrosis that does not conform to the pattern of either UIP or any other idiopathic interstitial pneumonia and for these cases the term ‘non-classifiable fibrosis’ is recommended.3 Table 6.1.2 summarises the pertinent features of UIP and the other interstitial lung diseases and may be used as part of a diagnostic algorithm.
Table 6.1.2 A diagnostic algorithm for usual interstitial pneumonia and the other interstitial lung diseases
| Collagenous or inflammatory? | Histology | Diagnosis |
|---|---|---|
| A predominance of collagen | Patchy subpleural and paraseptal distribution, loss of architecture, and presence of fibroblastic foci | Usual interstitial pneumonia (idiopathic pulmonary fibrosis in most cases) |
| With chronic peribronchiolar inflammation and possibly granulomas or isolated giant cells | Chronic extrinsic allergic alveolitis | |
| Stellate peribronchiolar scars (often masked by excess macrophages) | Burnt-out Langerhans cell histiocytosis | |
| No significant architectural disruption or consistent zonal distribution | Fibrotic non-specific interstitial pneumonia | |
| None of the above | Unclassified fibrosis | |
| A predominance of inflammation | Peribronchiolar with granulomas | Extrinsic allergic alveolitis |
| Peribronchiolar with eosinophils and Langerhans cells | Langerhans cell histiocytosis | |
| Peribronchiolar with intraluminal pigmented macrophages | Respiratory bronchiolitis | |
| Diffuse with hyaline membranes and fibroblast proliferation | Diffuse alveolar damage | |
| No consistent zonal distribution – mild intensity | Cellular non-specific interstitial pneumonia | |
| No consistent zonal distribution – marked intensity | Lymphoid interstitial pneumonia | |
| Non-necrotising granulomas in a lymphatic distribution | Sarcoidosis |
Aetiology and pathogenesis69,70
The histological features of IPF suggest that the disease results from repeated episodes of focal damage to the alveolar epithelium but provide no clue to the nature of the injurious agent. However, IPF has many features in common with the so-called collagen vascular diseases and the concept of lone versus systemic cryptogenic fibrosing alveolitis (CFA) links the two (see p. 264).71 Hyperglobulinaemia is common and circulating non-organ-specific autoantibodies such as rheumatoid factor and antinuclear factor are often present in IPF.61,72 In a national British survey of patients with ‘lone CFA’, rheumatoid factor was positive in 19% and antinuclear factor in 26%.16 Antibodies directed against the alveolar epithelium have also been identified.73,74 Immune complexes have been demonstrated in the serum75 and occasionally in the wall of pulmonary capillaries, particularly in early cases.76,77 It seems likely therefore that IPF has in part an autoimmune basis, with the alveolar epithelium being the target tissue (see Fig. 6.1.9). The initiating cause remains unknown although environmental and occupational pollutants,15,16,78,79 gastric reflux,80 viruses81–86 and drugs87 have all been incriminated from time to time. However, there is no firm evidence that any of these plays a causative role.88
Against IPF having an immune basis is the ineffectiveness of therapeutic immunosuppression3,23,33 and the accelerated rate of progression of the disease seen in the native lung of patients who have undergone single-lung transplantation despite the profound immunosuppression necessary to prevent rejection of the donor lung.89
The fact that the disease is occasionally familial19,20,90 suggests that genetic susceptibility may be important. There is evidence that in some families, susceptibility to the disease is inherited in an autosomal-dominant manner.90,91 One gene that is potentially responsible for such susceptibility is situated on chromosome 14q32, close to the alleles responsible for α1-antitrypsin deficiency.92 Rarely, several family members suffer from either IPF or α1-antitrypsin deficiency.93 Other genes identified include ones encoding for surfactant protein C and telomerase.94,95 HRCT of the asymptomatic relatives of patients with familial IPF identified abnormalities in 22%, with diverse patterns being found on lung biopsy.96 Several subtypes of idiopathic interstitial pneumonia may occur within the same family,90 which suggests that there may be an interaction between a specific environmental exposure and a predisposing gene (or genes) such that the insult predicates the pattern of fibrosing lung disease rather than the gene itself.
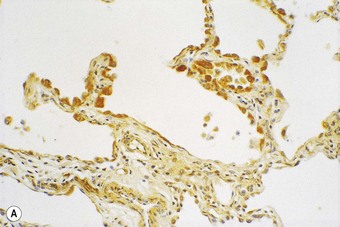
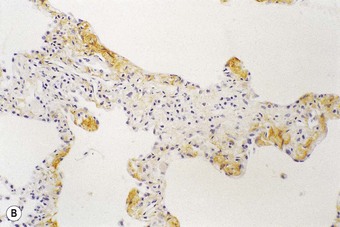
Following recognition of the alveolar epithelium as the site of initial injury, from which dysregulated repair and eventual fibrosis ensue, considerable attention has been paid to the cytokines associated with the regenerating epithelial cells. These cells express numerous profibrotic cytokines, including transforming growth factor-β (Fig. 6.1.11),97–99 interleukin (IL)-10,99 endothelin,100 tumour necrosis factor-α,101 insulin-like growth factor-1,102 transcription factor GATA-3,103 gremlin104 and connective tissue growth factor.105 Receptors for these profibrotic cytokines such as CCR7 have been localised to the interstitial compartment in UIP.106 Other factors identified in the regenerating epithelium include proteinases,107 tenascin98 and cytokines that inhibit cell migration and further re-epithelialisation, which may be important in the delayed or defective re-epithelialisation that has been held responsible for fibroblast recruitment, activation and sustained proliferation.108 Poor re-epithelialisation may be due in part to increased pneumocyte apoptosis, promoted by some of the above cytokines, for example, transforming growth factor-β.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


