Overview of Lymphoproliferative Disorders Associated with Primary Immune Deficiency Disorders
Keyur Patel, MD, PhD
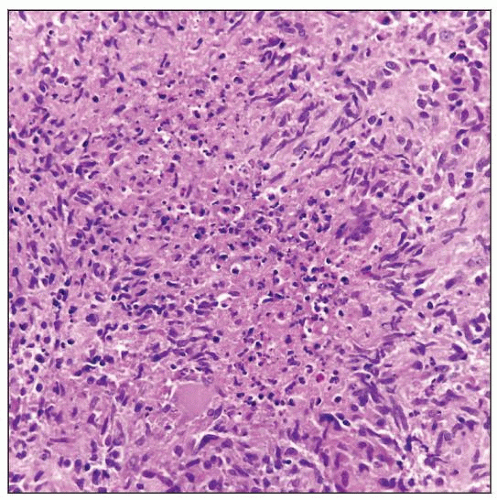 Chronic granulomatous inflammation involving lymph node in a patient with common variable immunodeficiency (CVID). |
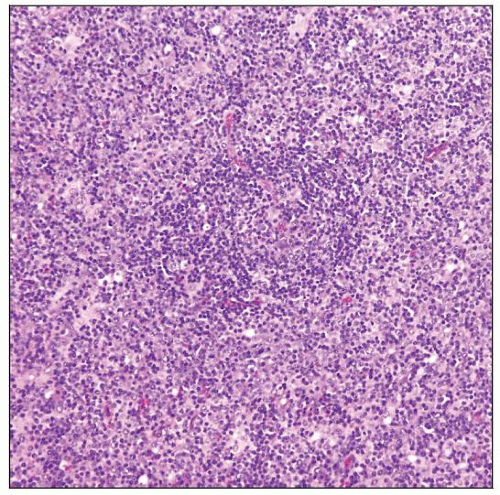 B-cell lymphoma with polymorphous features in a patient with ataxia-telangiectasia (AT). |
TERMINOLOGY
Definitions
Lymphomas and lymphoma-like lesions arising in clinical setting of primary immunodeficiency
Primary immunodeficiency disorders are a heterogeneous group of genetic diseases that result in an immunocompromised state
Abbreviations
Primary immunodeficiency disorders (PID)
Synonyms
Primary immune disorders
Congenital immunodeficiency diseases (disorders)
EPIDEMIOLOGY
Incidence
Variable incidence of clinically evident PID in USA
Cumulative incidence: 1 in 10,000
Incidence likely higher but some diseases are not evident clinically
Lymphoproliferative disorders (LPDs) associated with PID
LPDs are most common neoplasms in patients with PID
Up to 75% of all neoplasms in a given PID
Risk of developing LPD varies by type of PID, ranging 0.7-15% (accurate estimation is difficult due to low incidence)
Age Range
PIDs are more common in pediatric age group
Exception: Common variable immunodeficiency disease (CVID) occurs in adults
Median age of LPD onset: 7.1 years (per Immunodeficiency-Cancer Registry)
Recent increase in LPDs at older ages due to better survival of PID patients
Gender
More common in males; true for X-linked as well as for autosomal recessive disorders
ETIOLOGY/PATHOGENESIS
Etiology
Gene mutations account for many PIDs
X-linked hyper-IgM syndrome (XHIGM): CD40 or CD40 ligand (CD40LG)
Autoimmune lymphoproliferative syndrome (ALPS): FAS or FAS ligand (FASLG)
Ataxia-telangiectasia (AT): ATM
Nijmegen breakage syndrome (NBS): NBS1 (nibrin)
Complex abnormalities account for other PIDs
Wiskott-Aldrich syndrome exhibits defective function of T cells, B cells, neutrophils, and macrophages
Pathogenesis
Basis for increased risk of hematologic neoplasms is poorly understood; likely multifactorial
Epstein-Barr virus infection drives subset of LPDs in PID settings
Defective DNA mismatch repair involved in AT and NBS
Possible underlying immune defect against cancer cells
CLINICAL IMPLICATIONS
Clinical Presentation
Patients with PID often present with recurrent infection
Fever, fatigue, infectious-mononucleosis-like syndrome
Lymphadenopathy &/or hepatosplenomegaly occur in ALPS and X-linked lymphoproliferative syndrome (XLP)
In XLP, fulminant infectious mononucleosis (FIM) can occur, which is marked by fever, rash, generalized lymphadenopathy, and hepatomegaly
Site
LPDs in PID often present in extranodal sites
Treatment
Reduced risk of LPD after allogeneic stem cell transplant in PIDs
Limited data due to rarity of PIDs and lack of randomized trials
Recommendation is to treat with histologic subtype-specific protocol
Possible role for immunoregulatory therapy (e.g., interferon-α 2b)
Prognosis
Related to both underlying PID and type of LPD
Most LPDs in PID patients are clinically aggressive
Self limited, ALPS; clinically indolent, common variable immunodeficiency (CVID)
Newer antimicrobial therapies that allow more aggressive treatments have improved prognosis
Fatal hemophagocytic syndrome can occur in EBV-driven infectious mononucleosis
Occurs in XLP and severe combined immunodeficiency (SCID)
MICROSCOPIC FINDINGS
Nonneoplastic Lesions in Lymph Nodes
Spectrum of morphologic abnormalities
Subtle alterations may require immunophenotyping
Common findings
Lymphoid depletion
Atrophic follicles with progressive depletion of germinal centers
Depletion of small lymphocytes in paracortical region with increase in histiocytes and plasma cells
Similar findings observed in spleen and tonsils at autopsy
Secondary changes
Chronic granulomatous inflammation secondary to infections
Florid reactive hyperplasia
Atypical hyperplasia
Fatal infectious mononucleosis (FIM) resulting from EBV infection (XLP, SCID)
Extreme atypical hyperplasia
Polymorphous lymphoid cells with plasmacytoid and immunoblastic differentiation
Systemic uncontrolled proliferation of abnormal B cells
Frequent hemophagocytic syndrome, most readily identified on bone marrow aspirates
Waxing and waning lymphoproliferations (CVID)
Variable morphology showing follicular hyperplasia and paracortical expansion with many EBV(+) cells
Characteristic nodular lymphoid hyperplasia in gastrointestinal tract
Autoimmune lymphoproliferative syndrome
Expansion of CD4(-), CD8(-) T cells (so-called double negative cells)
Increased CD5(+) polyclonal B cells
Prominent follicular hyperplasia
X-linked hyper-IgM syndrome
Extensive accumulation of IgM-producing plasma cells in extranodal sites without malignant transformation
Peripheral blood B-cells express only IgM and IgD
Precursor Lesions
Broad morphologic spectrum
Increasingly dominant clonal population, from polyclonal, to oligoclonal, to monoclonal
Monoclonal expansions may or may not progress to major persistent lesions
Neoplastic Lesions
Increased risk of developing leukemias, lymphomas and nonhematopoietic tumors (lymphoma > leukemia)
In general, morphology and immunophenotype similar to lymphomas in immunocompetent hosts
Polymorphous cytologic features commonly seen
Non-Hodgkin lymphoma
Overall, B-cell more common than T-cell lymphomas
Exception: In ataxia-telangiectasia, T-cell lymphoma/leukemia is common
Diffuse large B-cell lymphoma (DLBCL) is most common NHL in PID patients
Immunophenotype similar to DLBCLs in immunocompetent patients
If EBV(+): Focal expression or absence of CD20 and CD79a; aberrant CD30(+)
Many cases are polymorphous with plasmacytoid differentiation
Frequently EBV(+)
Burkitt lymphoma is more common in XLP than in other PIDs
Hodgkin lymphoma (HL)
2nd most common LPD per Immunodeficiency Cancer Registry
˜ 10% of all lymphomas in PID patients
Classical HL most common in PID patients
Lymphocyte depleted and mixed cellularity types more common due to feeble immune response
HRS cells: CD15(+/-), CD30(+), pax-5(+, dim), CD45/LCA(-)
NLPHL relatively uncommon except in patients with ALPS
CLASSIFICATION
Immunodeficiencies
Combined T- and B-cell immunodeficiencies
Severe combined immunodeficiency (SCID)
X-linked hyper-IgM syndrome
Predominantly antibody deficiencies
Common variable immunodeficiency (CVID)
Other well-defined immunodeficiency syndromes
Ataxia-telangiectasia (AT)
Nijmegen breakage syndrome (NBS)
Wiskott-Aldrich syndrome (WAS)
Diseases of immune dysregulation
Autoimmune lymphoproliferative syndrome (ALPS)
X-linked lymphoproliferative disorder (XLP)
Congenital defects of phagocyte number, function, or both
Defects in innate immunity
Autoinflammatory disorders
Complement deficiencies
DIAGNOSTIC TESTS
Laboratory Tests to Diagnose PID
Multiple tests may be required to establish diagnosis of PID; however, testing for LPD in PID is same as in immunocompetent hosts
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


