- Cancer is a genetic disease characterized by the emergence of deranged versions of normal cells, born out of aberrant molecular biology.
- Cancer is the malign byproduct of an ensemble performance in which mutations in the DNA and altered gene expression are enacted against a background of conniving environmental factors such as carcinogens and chronic inflammation.
- A large number of factors are adduced to explain the genesis of cancer, including the twin pillars of incitement of primeval urges and the emancipation from normal restraining forces. Together, these produce untrammeled cell-cycle progression.
- Studies of rare familial “monogenic” cancer syndromes have had a major impact on our fundamental understanding of cell biology, but most cancers do not result from inheritance of single, potent, cancer-causing mutations.
- Instead, they are “sporadic,” with cancer-causing gene mutations arising in adult somatic cells.
- Hereditary factors may, however, exert weak and subtle influences on the risk of development and subsequently the behavior of most if not all so-called “sporadic tumors,” through a complex interplay between multiple, largely unknown polymorphic alleles, some of which may only be disadvantageous if the individual is exposed to particular environmental carcinogenic factors, such as tobacco smoke.
- In general, factors that cause mutations and those that increase cell replication can combine to cause cancer, which may explain the powerful role of chronic inflammation in the causality of many carcinomas.
- Cancer is a clonal disease arising by the multistep accumulation of genetic or epigenetic changes in tumor suppressor genes, oncogenes, and “caretaker” genes that favor expansion of the new clone over the old in a process akin to Darwinian evolution.
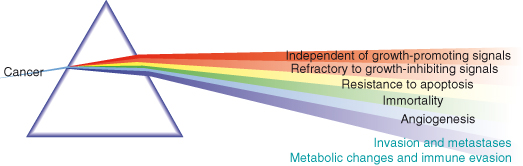
- Natural selection will favor expansion of clones with acquired characteristics advantageous to the cancer cells, often referred to as the “hallmark” features of cancer (Fig. 1.1), which have been famously distilled by Robert A. Weinberg and Douglas Hanahan as:
- Recently, the “Warburg effect,” a metabolic switch towards increasing ATP production by glycolysis, along with evasion or subversion of the immune system have been championed as the seventh and eighth hallmark features, respectively.
- RB and TP53, the doyens of tumor suppressor proteins, can arrest the cell cycle or trigger apoptosis in response to assorted cellular stresses, activation of DNA damage checkpoints, or during attempted oncogenic hijacking of cell-cycle control.
- An intriguing question is exactly how a tumor cell with DNA damage retains so many varied options in the face of TP53 activation? Thus, TP53 can mitigate cell death and inspire DNA repair but, in complete contrast, if repairs fail it might drive a cell to celibacy or suicide.
- Put simply, with respect to replication, a cell with irreparably damaged DNA has to either kick the habit or kick the bucket.
- Not surprisingly, therefore, loss of tumor suppressors is a prerequisite for tumorigenesis. Although there is some overlap, broadly speaking, cancer cells without TP53 can survive an alarming rate of mutation, whereas absence of the other archetypal tumor suppressor, RB, represents a fountain of youth for the otherwise rapidly senescing cancer cell.
- When and where, in the life history of a cancer, do the genetic changes required for metastases occur? There is no satisfactory answer to date. Natural selection does not really provide an explanation as to why a clone of cancer cells with metastatic capabilities would be selected for in the primary tumor, unless the causal mutations first and foremost also provide a growth advantage. It is possible that potential metastatic behavior is serendipitously acquired early in tumorigenesis as a byproduct of mutations promoting growth of the ancestral primary tumor (supported by some gene expression profiling studies of whole tumors). Alternatively, it may be that mutations in specific metastases-suppressing genes that do not confer a growth advantage to the primary occur at a later stage, possibly once cancer cells have begun circulating.
- Recent intriguing questions have been posed regarding the ongoing evolution of cancer cells in primary and secondary tumors. Recent findings suggest that following an initial shared origin, clones with metastatic capabilities emerge in the primary. Once ensconced within a new secondary environment, the metastatic alumni follow a parallel and distinct evolutionary path that may intriguingly begin while still in transit.
- Cancers are complex and heterogeneous, comprising a series of genetically differing populations (clones) of cancer cells. In fact, the dramatis personae of cancer includes the cancer cells-elect, the profligate parents, and a number of libertine relatives of dubious provenance.
- Moreover, the whole ensemble is supported by a strong supporting cast of both collaborating and insurgent noncancer cells that together constitute the cancer microenvironment.
- The cell of origin for any given cancer – be it stem cells that partially differentiate or differentiated cells that partially dedifferentiate, continues to offer opportunities for spirited debate.
- Tumors are not egalitarian societies. Rather they are in most cases oligarchies run by a malign minority of so-called cancer stem cells (CSCs). Part gang master and part queen bee, CSCs lie embedded within a large cast of bit part players. CSCs were first described in hematological malignancies, where they are strongly implicated in maintaining the malignant phenotype. More recently, CSCs have been identified in solid tumors and may be responsible for invasive behaviors, treatment resistance, and recurrence. By implication, these cancer oligarchs are the target of the original cancer-causing mutations, suggesting that in the case of a tumor the fish rots from the head.
- CSCs share many properties and molecular markers with normal stem cells, but this does not constitute proof of paternity. Under the influence of relevant mutations, including those that provoke epithelial–mesenchymal transition (EMT), normal cells can have “stemness” thrust upon them.
- This departs from the more traditional view of indefatigable clonal competition; dog eats dog, the strongest prevails with the extinction of the weakest – aut Caesar aut nihil.
- The cancer microenvironment, including the inflammatory milieu and the tissue stroma (connective tissue, fatty tissue, blood vessels, and lymphatics), represents an alma mater for cancer cells, which by encouraging EMT can help to generate CSCs and support the success of tumorigenesis.
- The greater recognition of the portentous events unfolding within the purlieus of the tumor peripheries during tumorigenesis has already yielded dividends. Thus, the stroma plays society hostess to a prohibition-free orgy of concupiscent cancer cells, egged on by a small faction of attendant immunocytes and under the averted gaze of the rest.
- Remarkably, it now transpires that cancer-contributing mutations are no longer the sole preserve of cancer cells themselves. In fact, mutations in stromal cells may allow them to more effectively mentor cancer cells towards the achievement of their six or eight hallmark milestones.
- The molecular profile of a tumor constitutes a manifesto, within which its future behavior is adumbrated and from which its weaknesses might be divined. Moreover, seminal parts of this manifesto achieve remarkably widespread circulation. Therefore, for diagnosis it may be unnecessary to directly remove tumor tissue, because cells, proteins, and nucleic acids derived from it are continually being shed into more readily accessible body fluids.
- The search for clinically useful molecular biomarkers represents one of the most promising areas of cancer research. Many biomarkers are already in routine clinical practice, where they assist in disease monitoring and in treatment selection.
- However, biomarkers have, as yet, not helped us to paint more accurate portraits of tumors. Unfortunately, in most cases they fail to unambiguously identify their subjects. There is no “Habsburg lip” for cancers.
- In fact, biomarkers have had limited impact on screening the general population for most cancers.
- Given the increasing number of therapeutics in our arsenal against cancer, great efforts are being made to find biomarkers that may help select appropriate treatments for individual patients.
- Cancers may be cured by surgery, but only if the entire tumor is accessible and no cells have spread to other sites. Modern approaches to cancer drug development are increasingly moving away from traditional chemotherapeutic agents which paralyze cell division or cause DNA damage and instead are aimed at targeting specific cancer-relevant proteins, such as oncogenic tyrosine kinases.
- Oncogene addiction, the process by which cancer cells become critically reliant on the mutant signaling molecules, offers the potential of both effective and minimally toxic agents directed against such proteins. A potential realized by pioneering therapeutic successes, such as imatinib, used to such good effect to target the abnormal BCR–ABL fusion protein in chronic myeloid leukemia.
- However, use of these agents is in most cases severely limited by acquired or, on occasion, inherent resistance of cancer cells to the treatment. It is hoped that understanding the resistance mechanisms involved will allow rational development of combinations of targeted agents in the future, though further mutations may render even these ineffective over time. One could easily be forgiven for likening these efforts to cure cancers by drug therapy with the task set before Sysiphus.
- However, we may yet keep the boulder from rolling down the hill. Knowledge is power and by exploiting the potential of treatment biomarkers we may gain an edge over cancer. Thus, we can assess whether a given cancer will respond to particular drugs as exemplified by the presence of estrogen or progesterone receptors and mutant NEU in breast cancer, or may conversely suggest a response to be unlikely as in the presence of KRAS in colorectal cancer. Armed with enough of these biomarkers there is reason to suppose that the goal of individualized medicine and tailored therapy may soon be within reach.
Introduction
And yet there is something so amiable in the prejudices of a young mind, that one is sorry to see them give way to the reception of more general opinions.
Jane Austen
In this chapter we give a historical overview of cancer and go on to introduce and summarize the concepts and topics to be covered in this book. Wherever possible, we emphasize new thinking, emerging views, and novel models for studying and understanding oncogenesis. Unapologetically, this chapter aims to be stimulating and thought-provoking.
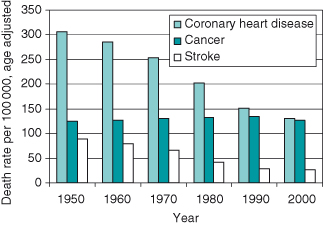
Cancer has been recognized throughout recorded history and was known to the ancient Egyptians (see Appendix 1.1 – History of cancer), but it was not until the seventeenth century that the formal study of cancer (oncology) was first documented. As with much of biology, the last 50 years has witnessed spectacular progress in describing the fundamental molecular basis of cancer following the advent of molecular biology and genetics. Frustratingly, such exponential progress in describing the biology of cancer has not yet translated into an equally impressive progress in the war against most common cancers (Fig. 1.2). We can, however, claim victory in some important skirmishes. Possibly the single greatest success has been in altering the status of cancer in many cases to that of a chronic illness. Most people now live for some time with the disease rather than rapidly succumbing to it. This is in part testimony to better treatments. At the time of going to press, the overall median survival time for the commonest 20 cancers had increased from around one year in 1971 to just under 6 years by 2007. Most of this gain, however, reflects the very pronounced improvements in survival from breast and colon cancers and from lymphoma. Unfortunately, over the same period, for other common cancers, notably those of lung, pancreas, and brain, improvements have been negligible and survival time is still measured in weeks.
Figure 1.2 The incidence of cancer is not declining when compared to other major diseases, yet in the United States alone more than US$4.7 billion per year is spent on cancer research. Leland Hartwell and others at a meeting of the American Association of Cancer Research identified the following areas, in addition to developing new therapies, as key targets to address this major public health issue: (1) More coordinated and concerted activity between researchers. This would require establishing the necessary infrastructure for facilitating collaborative working and information exchange. (2) Testing drugs and agents in early-stage disease rather than as at present largely in end-stage cancer (we may be underestimating the potential of many drugs and therapies for this reason). (3) Real-time monitoring of treatments in early-stage cancers, though to identify earlier stages will require improved biomarkers. (4) Use of RNAi to explore combinations of targets. (5) Improved understanding of chromosomal aberration. This occurs very early in mouse tumors. (6) Exploiting genomic instability in therapeutics. Understanding more about DNA repair and repair of double-strand breaks (the latter are unusual in mouse unless telomeres are shortened). (7) Improved diagnostics from blood and body fluids – proteomics (less than 1% of proteins in blood identified, and less than 20% of these licensed for diagnostics).
Data from Centers for Disease Control.
At first glance, the biology of cancer appears straightforward. Cancer cells stop obeying the “societal” restraints imposed on individual cells within the adult organism. Instead, they multiply uncontrollably and congregate in places that should be off-limits – much like teenagers. However, in order to achieve such independence cancer cells must first be emancipated and overcome the numerous intrinsic and extrinsic barriers that seek to prevent such selfish behavior before it can threaten the survival of the entire organism.
In this book we describe the means by which normal cells are transformed into cancer cells and the key cellular processes subverted along the way. We also describe the cellular forces arraigned against the designate cancer cells and those operating on behalf of them and the weaponry available to both sides. We explain how this basic knowledge has been translated into improved diagnostics and more biologically targeted therapeutics for cancer patients. The global burden of different types of cancer is described alongside the current state of the art in diagnosis and management of cancer patients. Along the way, we make some predictions as to where new scientific and clinical breakthroughs may come from and offer our humble opinions as to why, despite some notable successes, cancer cells continue to flourish even in the face of our most sophisticated anticancer therapies. Maybe we should accept at this point that perfect theoretical proof of fact is impossible. The Münchausen Trilemma (used in philosophy to imply that it is not possible to prove any truth, even in mathematics) may provide some reassurance on this point. However, in practical terms, a good model incorporating treatments or biomarkers that work in the clinic may be a more realistic goal, even if our understanding of why the treatments work proves misguided. We are sure to continue to use the treatments until something better comes along, even if we discard the model.
Cancer poses a major threat to already overstretched healthcare services. The magnitude of the problem was summarized by Dr Gro Harlem Brundtland, former Director-General of the World Health Organization, in the Foreword to the 2003 World Cancer Report: “The global burden of cancer continues to increase. In the year 2000, 5.3 million men and 4.7 million women developed a malignant tumour and 6.2 million died from the disease. The number of new cases is expected to grow by 50% over the next 20 years to reach 15 million by 2020.”
Cancer is responsible for more than 10% of deaths worldwide and more than 25% in some countries. Excluding the relatively frequent nonmelanoma skin cancers, lung cancer is the commonest cancer worldwide, accounting for 1.2 million new cases per year, followed closely by breast cancer and colorectal with around 1 million new cases.
The high incidence of this disease, its life-threatening nature, and often unsatisfactory management has motivated academic researchers and those from the biotechnology and pharmaceutical industries to focus on the causes and potential treatments of cancer on a scale unparalleled in almost any other disease area. Remarkably, at present there are almost 500 products in clinical trials, of which 100 are in phase III, with breast cancer and non-small-cell lung cancer receiving the most attention.
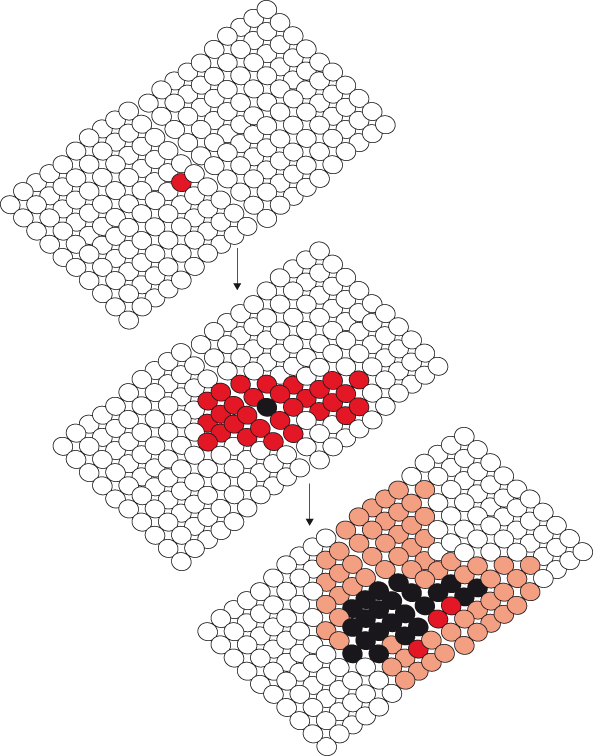
In general, cancers begin with a mutational event in a single cell and then develop in multiple stages through the acquisition of further mutations, propitious and otherwise, that are passed on to the progeny of that cell when it divides. So cancer is a clonal disease (Fig. 1.3). Aside from a few notable rare exceptions, these events arise predominantly in adult somatic cells and so are not inherited by the offspring of the affected cancer patient but only by the progeny of the affected cancer cell. In other words, transmission of the mutation ceases with the death of the patient, unless by some chance the mutant gene has been picked up by a virus, which survives and propagates. If such a virus carrying a mutated gene infects a potentially vulnerable host then the cancer-causing potential of that gene may again be unleashed upon another hapless organism. Contrary to accepted wisdom, very recent studies have suggested the astonishing possibility that cancer cells could under rare circumstances be directly inoculated from a tumor-bearing host into an unfortunate recipient. Thus, leukemias may be transmitted from mother to child and dogs may transmit cancer cells to their partners during mating.
Figure 1.3 Cancer is a clonal disease. Expansion of the original clone (red) is followed by emergence of a new clone (black) which gradually replaces the original. Subsequently, a further clone (orange) emerges and expands.
Mutations – alterations in the coding sequence of the DNA – are not the only route to inactivation or activation of a key gene/protein. Gene expression may also be strongly influenced by a variety of epigenetic factors that alter chromatin structure without changing the coding DNA; these can still be passed on through successive cell generations. The term “epimutations” is often used to encompass both these major routes by which cancer cells acquire aberrant expression/activity of key genes and proteins. The average adult human has been estimated to contain as many as 1014 cells (i.e. 100 000 000 000 000 cells), most of which could theoretically become a cancer cell given the right sort of genetic mutations and epigenetic changes. In fact, cancer is unique in that epimutations in a single cell can give rise to a devastating disease because the resultant aberrant gene and associated antisocial behavior are transmitted to all the cellular progeny of that cell.
Because DNA replication and synthesis are essentially error prone, it is replicating cells that are most vulnerable to cancer-causing mutations. Not surprisingly, as stem cells are the main replicating cell population in the hematological system and also in epithelia, from which most cancers arise, they have long been intimated as the cell of origin for cancer. This is supported by the presence within many cancers of a side population of cells bearing stem cell characteristics known as the “cancer stem cells” (CSCs). More of this later. Although some differentiated cell types, of which adult nerve cells are a good example, are by implication unlikely to give rise to cancers because they are essentially nonproliferating in the adult, most cells either regularly replicate or can do so at a pinch. Most adult cells survive on average for 4–6 weeks and then have to be replaced. Over a hundred billion cells may die each day and are renewed either by replication of existing cells or from stem cell precursors. Given that each cell gets a substantial amount of daily DNA damage and 1011 or more of them will replicate each day, that is a lot of potential cancer cells!
With this in mind, a cancer might be expected to be a frequent occurrence. Yet cancer is diagnosed in only in 1 in 3 people and usually even then only after 60 or 70 years of potentially mutation-causing events. So why does a clinically apparent cancer only arise in every third individual when there are somewhere in the region of 1014 good potential cellular targets at risk? Moreover, we live in a world in which each of those cells is continually exposed to a myriad of avoidable and unavoidable DNA-damaging agents. Let us state this in the boldest terms possible. At a cellular level, cancer is very, very rare. This surprising observation can only be accounted for by the existence of some extraordinarily effective barriers to cancer cell development. These barriers are clearly not infallible, but they must be tremendously powerful. It is also worth reflecting on what the purpose of many of these conserved anticancer mechanisms may have originally been, given their presence in short-lived and even oligo-cellular “organisms” that are at no risk of cancer. One possibility is that processes such as senescence and apoptosis are a byproduct of archaic processes involved in balancing nutrient supply, growth and repair, and energy that fortuitously also limit cancer in longer lived multicellular organisms.
Cancers may well originate from a single bad cell, but are self-evidently not clinically detectable at that stage either by direct observation or conventional investigations such as X-ray,. This requires the presence of a small nodule, at which point replication has increased the number of cancer cells to around one billion (109). In other words, by the time a cancer is discovered, the original cancer cell has proceeded through some 30 or more cell divisions, and acquired a host of further epimutations. This situation is compounded by the near universal loss of normal DNA damage surveillance and repair processes. Not surprisingly, this has complicated studies attempting to unravel the initial causes of cancer in humans. We are detectives investigating a crime that occurred some 30 generations in the past. Imagine today trying to identify the particular something, potentially quite innocuous, that happened to one of my antecedents at the time of the battle of Hastings, something which may in some unexplained way have been propagated through the ages and finally made me write this book.
Safely squirreled away within a stromal nursery, those normal cells that have been successfully emancipated will give rise to a new dynasty of proto-cancer cells. This new found freedom may result from chronic inflammation but once rendered immutable by epimutations a door to a malignant future has been forced open. The resultant unfettered cell can indulge in previously proscribed behaviors, such as unrestrained growth, and be afforded unprecedented opportunities for travel and preferment. Gradually, successive generations, honed by exposure to the hostile forces arraigned against them, will witness the emergence of increasingly malign elites that begin to dominate and supplant their forbears. If we can stretch the societal analogy further, then we may claim that normal tissues exemplify totalitarianism, whereas cancerous ones are essentially pluralist. Although with time, one or more clones may become first among equals and even have imperialist aspirations, it is now recognized that in many cases tumors continue to harbor substantial representation of earlier clonal dynasties.
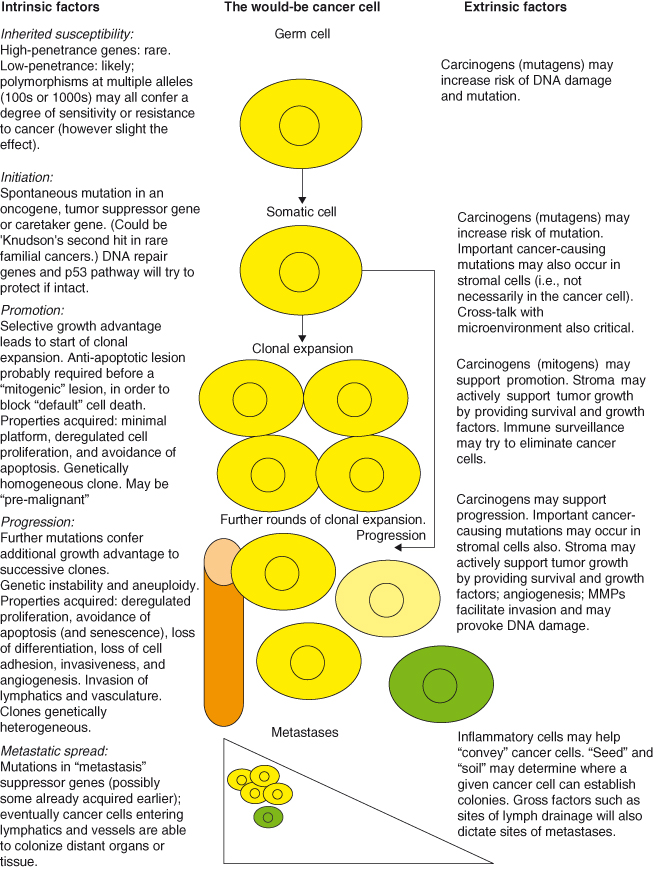
A schematic of how we believe cancers arise and progress is provided in Fig. 1.4. This, can be used as an overview to be referred to while reading the more detailed (and complex) description of the basis of cancer in this book.
Figure 1.4 A highly stylized potential “life history” of a cancer cell. Cancer cells are shown in yellow (different shades denote subclones); stromal cells are green, vessels are orange.
Cancer Incidence and Epidemiology
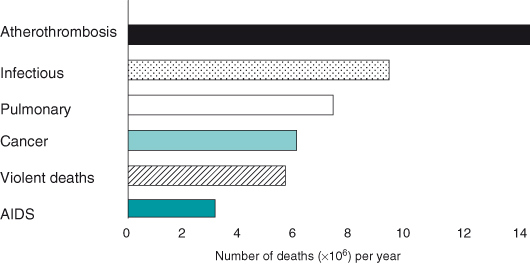
In the United Kingdom and North America, the lifetime risk of developing cancer is more than one in three, and cancer is responsible for around one in four deaths. Yet, the fear of cancer experienced by many individuals should be balanced by an appreciation that one is still far more likely to die or become disabled due to a heart attack or stroke (Fig. 1.5), if that knowledge may be in any way regarded as reassuring.
Figure 1.5 Leading causes of death worldwide, 2004.
Redrawn from Murray CJL, Lopez AD (1997) Mortality by cause for eight regions of the world: Global. Burden of Disease Study. Lancet, 349: 1269–1276.
Given that almost every cell type can give rise to cancer and that more than 200 different types of cancer are recognized, it is notable that four – breast, lung, large bowel (colorectal), and prostate – account for over half of all new cases. It should also be noted that although nonmelanoma skin cancer (NMSC) is very common, with 100 000 new cases recorded each year in the United Kingdom, this data is likely incomplete and the disease usually curable, so the NMSC statistics are now routinely omitted from the overall totals. In 2006 in the United Kingdom, 293 601 people were diagnosed with cancer, excluding NMSC.
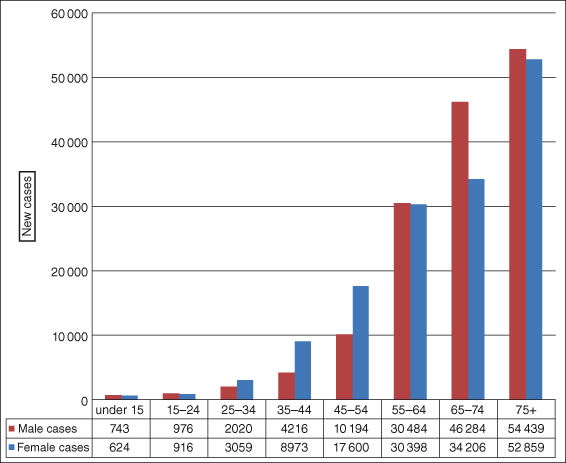
Different cancers affect people at different ages, but not surprisingly the overall risk of developing a cancer rises sharply with increasing age, with 65% of cancers in the United Kingdom occurring after the age of 65 years and 35% above age 75 (Fig. 1.6). In children, leukemia is the most common cancer (around 30% of all pediatric cancers); in young men aged 20–39 it is testicular cancer.
Figure 1.6 Cancer development by age for 2007 in the United Kingdom.
Modified from International Agency for Research on Cancer (IARC) and Cancer Research UK data
.
The incidence of cancer has changed over the last 20 years; there has been a decline in lung cancer in the United Kingdom and North America in men (but an increase in women), mainly as a result of changes in smoking habits, and an increase in breast and prostate cancer. Yet despite this, an estimated 160 000 people died from lung cancer in the United States alone in 2009. In 1981 there were 78 cases of breast cancer per 100 000 women in Great Britain, and 38 cases of prostate cancer per 100 000 men. By 2009 rates were 124 and 106, respectively (http://info.cancerresearchuk.org/cancerstats/incidence).
The International Agency for Research on Cancer (IARC) has released figures on global cancer incidence for 2008 and made predictions for the next decades (http://globocan.iarc.fr). Globally, around 12.7 million new cases and 7.6 million cancer deaths occurred in 2008, the commonest being lung (1.6 million), which makes up almost 13% of the total, breast (1.38 million), and colorectal cancers (1.23 million). The most common causes of cancer death were lung, stomach, and liver, indicating the relative success of treatments for breast and colon cancers.
It has long been appreciated that there is a geographical variation in cancer incidence and deaths. Importantly, 56% of cases and 63% of cancers and deaths were in the developing world. Of the estimated 371 000 new cases of cervical cancer in 1990, for example, around 77% were in developing countries. This latter case likely reflects socioeconomic pressures and the prevalence of causal factors such as certain strains of the human papilloma virus (HPV). Globally, the most common cancer affecting women is breast cancer, followed by cervical cancer. However, in North America the most common cancer in women after breast cancer is lung or colorectal. Around 226 870 women are predicted to develop breast cancer in the United States in 2012 and around 226 160 men and women will develop lung cancer, and around 143 460 colorectal cancer (www.cancer.gov/cancertopics/commoncancers). The data for this period should soon be available. Predictions for global cancer make sobering reading: it is predicted that by 2030 there will over 21 million new cases and above 13 million deaths each year from cancer.
Race and gender also influence rates of cancer and this is graphically illustrated by data from 1999 from the United States Department of Health and Human Services. Some of the findings, such as lower rates of melanomas in men and women of Afro-Caribbean origin, attributed to inherent protection from UV exposure, are predictable. Others, however, are less so. Thus, although prostate cancer is the most frequent cancer in males, rates are 1.5 times higher in Afro-Caribbean men than in white men. Similarly, the leading cancer in women, regardless of race, is breast cancer, followed by lung/bronchus and colon/rectal in white women and colon/rectal and lung/bronchus in Afro-Caribbean women. Breast cancer rates are about 20% higher in white women. Multiple myeloma and cancer of the stomach are among the top 15 cancers for Afro-Caribbean women but not white. Recent data have become available for the United States from 2005, which shows that the rate of all cancers combined for black, white, Hispanic, Asian/Pacific islander, and Native American Indian are 591, 526, 406, 314, and 280 thousand per annum respectively (http://apps.nccd.cdc.gov/uscs/).
Towards a Definition of Cancer
A definition of poetry can only determine what poetry should be and not what poetry actually was and is; otherwise the most concise formula would be: Poetry is that which at some time and some place was thus named.
Karl Wilhelm Friedrich Schlegel
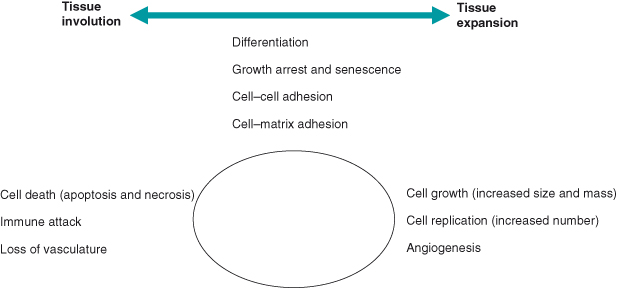
The terms “tumor” or “neoplasm” are used interchangeably to describe a diverse group of conditions associated with uncontrolled cell replication. Tissue mass is normally tightly controlled to serve the needs of the organism. This control is achieved by the balancing of various and often opposing cellular processes (Fig. 1.7). Disturbing the balance of these processes results in diseases; if cell losses exceed renewal this results in degeneration/involution, whereas the converse results in tissue expansion, hyperplasia, or neoplasia. If the expansion in cell numbers is confined locally then it is described as “benign,” but if this unscheduled cell replication is accompanied by invasion of surrounding tissues or spread to distant sites (“metastasis”), then it is unambiguously described as malignant.
Figure 1.7 Processes contributing to regulation of tissue mass. Cell mass is determined by the balance of various cellular processes including, at the two extremes, growth/replication and cell death.
These terms are relatively straightforward as they are descriptive and based on gross observations. It should be remembered, however, that the pathological definitions of benign and malignant do not always translate into similarly benign or malign outcomes for the patient. Thus, a benign brain tumor causing severe neurological disturbance may be inoperable or require potentially life-threatening surgery, whereas a malignant prostatic cancer or microscopic metastases may have had no clinical impact and be discovered accidentally at post mortem. Adenomas are benign tumors originating in glandular or secretory tissues (such as lactotroph adenomas of the pituitary, which secrete prolactin, or parathyroid adenomas, which secrete parathyroid hormone – PTH). Such adenomas can result in substantial morbidity as a result of deregulated secretion of hormones and may also progress to become malignant, when they are termed “adenocarcinomas.”
Classification of cancer
Classification of cancer is complicated by the variety of human cancers, with hundreds of different tumor types arising from almost every tissue and in every organ. This is further complicated by the ability of a cancer cell to invade surrounding tissues and metastasize to distant organs. Cancer biologists and oncologists have agreed on a classification based on the tissue of origin, regardless of organ location, focusing on the similarities in cellular structure and function among these tumors. Tumors are generally classified as either liquid or solid. The former includes leukemias and lymphomas comprising neoplastic cells whose precursors are usually motile. Solid tumors comprise either epithelial or mesenchymal cells that are usually immobile. Pathologically, cancers are classified as:
- carcinoma, originating from epithelial cells in skin or in tissues that line or cover internal organs and typically represent over 80% of human cancers;
- sarcoma, originating in bone, cartilage, fat, muscle, blood vessels, or other connective or supportive tissue;
- leukemia, a cancer originating in blood-forming tissues, such as the bone marrow, causing large numbers of abnormal blood cells to be produced and enter the bloodstream; and
- lymphoma, originating in the cells of the immune system.
It is worth emphasizing that the purposes behind disease classification are to help make the most accurate predictions about prognosis and response to particular therapies in the clinic and in the laboratory to ensure that as far as is possible like is studied alongside like. As discussed later, this objective may increasingly be better served by grouping cancers on the basis of their shared molecular pathoetiology rather than by tissue of origin.
“Carcinoma in situ” refers to lesions regarded as cancer that remain localized to the tissue of origin, often constrained by intact basement membrane. Such tumors often respond well to treatment, with good prognosis for the patient. In contrast, “invasive carcinomas,” by disrupting basement membranes and growing into surrounding tissues, are more difficult to treat successfully. In addition, since invasion is usually a prerequisite for metastasis, the ultimate cause of most cancer-related deaths, even when the local lesion is treated, the prognosis is often poor.
Importantly, disease classification is not written in stone, as technical advances are made and larger numbers of individuals with a given disease are studied, it is often possible to recognize previously unappreciated “subclasses” of disease that can readily be detected and further improve accuracy of prognosis and prediction of treatment responses. Most recently, advances in postgenome era technologies such as oligonucleotide arrays and proteomics (Chapter 20) are allowing a subclassification of cancers in terms of molecular profile termed “tumor fingerprinting.” At the same time genomics (essentially reading the DNA) is being increasingly applied to look for cancer susceptibility genes in patients and for mutations in tumors. It is hoped that in the future such powerful tools will ultimately allow more accurate determination of prognosis and even “tailored” therapy, whereby each patient can be uniquely classified and treated on the basis of such tests. These aspirations are often referred to as “individualized medicine”, reflecting the ideal of being able to treat each individual in a uniquely appropriate way, based on variation in one or more of the following parameters: gene alleles, gene expression/protein expression and mutations in tumor cells, proteins in the blood.
It Is Surprisingly Difficult to Define Cancer in Practice
Cancer is a difficult term to define accurately. Put simply, cancer is synonymous with malignancy, and refers to a group of conditions that have manifested malignant behavior, namely unscheduled and uncontrolled cell growth leading to invasion and/or metastases. There is no ambiguity in this case as the definition is “retrospective” and based on the readily observable behavior of the “cancer.” Such a narrow definition is of limited practical value in the laboratory, however, and particularly in the clinic, as it precludes true preventative or even early treatment. This seemingly abstract issue is placed in context when it is remembered that for those cancers where rates of death have actually been reduced over the last few decades, this has resulted primarily from improvements leading to earlier diagnosis and earlier administration of treatment.
It is clear that certain features at a microscopic level can accurately be employed to identify a tumor as cancer before it manifests overtly malignant behavior clinically (metastasizes to lymph nodes or other organs or has on imaging or surgery been shown to have invaded local structures). In other words, a cancer is a cancer before it necessarily declares itself by behaving as one. In most cases, this requires the demonstration of evidence of penetration of a basement membrane or invasion into surrounding tissue (which means you need to look at a piece of tissue that includes the tumor – histological examination) and/or the presence of “cancer cells,” namely cells exhibiting defined changes, which from experience are the same or similar to those seen in circumstances which are incontrovertibly cancer (which means you need to have acquired some cancer cells from body fluids, sputum, or via a smear- cytological examination). Clearly, the latter is often quicker and less invasive in clinical practice.
In a clinical setting, where the primary purpose is to identify a tumor or lesion that requires surgical excision or other treatment, it may be sufficient to know that a particular lesion (based on gross appearance or histological examination) poses a risk of proceeding to an invasive cancer. A lesion may already be regarded as a cancer, on the basis of abnormal growth or appearance and the near inevitably of progression to invasion (carcinoma in situ), or its potential may not be yet realized/manifested but risk of progression is high (“precancerous” or “premalignant”). This forms the basis for identifying “high-risk” lesions such as breast carcinoma in situ, Barrett’s esophagus (a precursor of esophageal cancer), colonic polyps (a precursor of colon cancer), and others. Cytological examination may identify premalignant cells and is employed where such cells can readily be obtained, including cervical screening for the early detection and prevention of cervical cancer (Box 1.1).
For a research scientist, these distinctions are also of critical importance. The ability to define the point at which a premalignant benign lesion ends and a malignant cancer begins is a prerequisite to understanding the initiation and key early events in cancer formation. In the laboratory the progressive behavior of transformed cells or tumor progression can be investigated in animal models, as long as the necessary investigative tools are available, but this opportunity is self-evidently usually lacking in the study of cancer in humans. The cancer researcher can validate predictions made about the future behavior of a given lesion by prospectively tracking the eventual emergence of invasive metastatic cancer but, as will become clear later, the actual stage of evolution at which cancer cells emerge and acquire ability to become invasive and metastasize is still contentious and quite difficult to detect.
Cancers May Not Always Be Clinically Apparent
Difficulties of definition notwithstanding, the clinical situation is further complicated by the increasing awareness that microscopic colonies of cancer cells (in situ tumors) can be detected in different tissues (thyroid, breast, prostate for example) at autopsy in most older individuals. In fact, such clinically irrelevant in situ cancers may be a 100- to 1000-fold more common than clinically apparent cancers arising in those same tissues during life. For example, most older individuals have in situ thyroid carcinomas at autopsy, whereas only around 0.1% of similarly aged individuals are found to have thyroid cancer during life. Although biologically intriguing and testifying to the potential effectiveness of innate anticancer defenses (such as antiangiogenic factors), such findings may increasingly be problematic in the clinic.
Until recently, we have generally not detected the vast majority of such in situ tumors during life, largely because we do not routinely biopsy tissues in apparently healthy individuals. However, one area in which detection of such in situ tumors may pose difficult and as yet unresolved clinical dilemmas, is increasing use of diagnostic prostatic biopsy in older men, and discovery of so-called “incidentalomas” during routine imaging procedures such as CT and MRI scanning. The now ubiquitous presence of privately run “walk-in” imaging centers offering the dubious benefits of whole-body scans will undoubtedly compound this problem. For example, what do you do about the incidental lump that is not self-evidently cancerous – particularly as benign irrelevant lesions will be considerably more numerous? The patient will likely be anxious and may well push to undergo potentially dangerous invasive diagnostic steps in order to be as certain as possible that they do not have cancer. Guidelines have had to be developed and will continue to be needed to assist clinicians in deciding which individuals with such findings actually require any form of treatment or just reassurance.
However, this is far from straightforward, as there are many cases where the actual ability to predict the risk of future invasive cancer based on the appearances of a given lesion are not yet sufficiently mature. A good example is the readily visible dysplastic white lesion in the mouth that may in some cases – but by no means all – herald the development of an oral squamous cell carcinoma. Ironically, at least in some cases, where the lesion may be less likely to come before the eagle eyes of dentists and GPs or it is not technically possible to detect the early lesion let alone examine it, this may be for the best until our ability to more accurately predict the future behavior of these early lesions improves and/or we greatly increase our current arsenal of sufficiently well-tolerated and nonharmful therapies to exploit the potential benefits of early diagnosis.
This interesting debating point notwithstanding, it is abundantly clear that in order to prevent or cure cancer effectively it is essential to diagnose disease as early as possible, and nothing should distract us from our efforts to progress in this goal. Failure to do so will inevitably mean that potentially life-saving early treatment for some individuals destined to develop clinically important cancer will be delayed. To resolve this conundrum is theoretically simple – we just need to distinguish early lesions that will never progress to disease from those that will progress to cancer. Although, routinely screening apparently healthy individuals for certain cancers has been well-validated and has become accepted best practice for cancers of breast (mammography), cervix (Pap smear), and colorectum (fecal occult blood) in many countries, for most cancers we urgently need better tests and tools. Fortunately, the research community has responded to this challenge and much progress is being made in finding new tests and “biomarkers” for various cancers that might give important information about prognosis and treatment response (see below and Box 1.2).
As mentioned earlier, in order to improve our predictive/diagnostic abilities, traditional examination of patients in the clinic, application of imaging techniques, and cytology/histology of the tumor are increasingly being supported by newer techniques, such as molecular profiling. Traditionally, genetic analysis looks for single susceptibility genes that confer a high risk of cancer formation, but in future this may include more complex genomic testing (of multiple polymorphic alleles – see below), or direct analyses of gene/protein expression in the tumor by various techniques including gene chip microarrays and proteomics. Considerable enthusiasm has been generated by the possibility of using relatively noninvasive tests to identify cancer biomarkers in blood samples or other body fluids from patients with cancer or at risk of cancer. Thus, proteins, mRNA, or miRNA derived from the tumor or from the body’s response to it might be analysed in body fluids. In many cases it has also proved straightforward to isolate and examine cancer cells (or their DNA) from blood or topically. If such information can be correlated with the presence or absence of cancer in the healthy population, or with clinical outcome or treatment response in known cancer patients, then these will be useful biomarkers.
The best-known currently used serum biomarker is prostate-specific antigen (PSA), elevated levels (or progressively rising levels) of which are associated with significant risk of prostate cancer. However, this falls short of the ideal in several respects, in particular the number of false positives (the test wrongly suggests the possibility of prostate cancer) and false negatives (a cancer fails to be diagnosed). This means that even clinical trials disagree on the benefits of general screening with PSA. In fact, there is a more fundamental flaw in the notion of simply detecting presence of prostate cancer by screening: it gives no insights into prognosis. This quandary is easiest to appreciate if we assume an ideal performance for the test and thus have in some way eliminated false positives (without compromising sensitivity). So now we use the test and it unambiguously tells us which patient has prostate cancer. What it does not tell us is what to do with the patient. Why not? Because recent trials have suggested that PSA screening results in overtreatment because the prognosis of occult prostate cancer is so variable and often does not affect mortality or morbidity (see Chapter 3). This does not mean that PSA screening is without value, it makes a major contribution to the investigation of patients with symptoms of prostatic enlargement (difficulty in micturition) and in the follow-up of prostate cancer patients following treatment. However, how suitable PSA is for screening the general population is controversial.
Tumor-derived biomarkers are already in routine use in the clinic. Thus, the presence of estrogen and progesterone receptors or of a HER2 mutation in breast cancer defines patients who will likely benefit from hormone-based therapies or trastuzumab, respectively. Commercial biomarker assays which measure expression of multiple genes and mutant versions from tumor samples by reverse transcription polymerase chain reaction (RT-PCR), such as the Oncotype DX test for breast cancer (measures HER2, ER, and PR status as well as 13 other cancer-relevant genes including Ki-67 and survivin) are now available. The presence of mutant KRAS in a colorectal tumor identifies a subgroup who will respond poorly to drugs targeting epidermal growth factor receptor (EGFR).
Genotype may also be helpful. Thus, a recent large study has confirmed that breast reduction surgery in women who are carrying germline mutations in BRCA1 and BRCA2 can markedly prevent breast cancer in these individuals. In a recent study, there were no diagnosed cancers in 247 women with risk-reducing mastectomy compared with 98 women of 1372 diagnosed with breast cancer who did not have risk-reducing mastectomy. Moreover, women undergoing risk-reducing salpingo-oophorectomy had improved survival.
If We Find a Cancer What Do We Do with It?
Not only do we often not know who to treat, we are often unsure what treatments to use, particularly before the development of an obviously life-threatening cancer. This situation has not been helped by the fact that the majority of therapeutic trials have focused largely on the end stages of cancer, by definition, the point at which these therapies are least likely to successfully cure the disease. Why? Because regulatory approval requires a lengthy series of clinical trials (see Chapters 15 and 16) and these are most readily conducted in patients with advanced cancer for whom no further treatments are available. Use earlier in the disease process is often left until after marketing and then interpretation may be confused by the need to use the new drug alongside existing best practice.
Improved ability to predict treatment response is fundamental to avoiding the morbidity and mortality associated with cancer while also restricting potentially harmful or even life-threatening treatments to those individuals most likely to benefit. Most treatments are justifiable when a life-shortening cancer is prevented, but would be very undesirable if employed in an individual never destined to develop cancer and who will eventually die of some unrelated other cause and whose life would have been affected less by the cancer than by the treatment. In practice, what is needed are clinical measures or new biomarkers that correlate with prognosis and that ideally also assist in selecting the best treatment or combination of treatments (from among watchful waiting, surgery, radiation, and drugs).
One thing is clear: early treatment offers the best chance of a successful outcome. This problem is addressed by various screening programs aimed at identifying premalignant or early stage cancers (see Box 1.1). Importantly, in these cases suitable treatment strategies have been defined.
As discussed in the previous section, it is hoped that detailed molecular analyses of tumor samples or body fluids will not only improve our understanding of the “roadmap” to cancer for any given cancer, which might in turn guide us to the application of specific drugs to target particular genes/proteins, but may also improve our ability to predict therapeutic responses. Such detailed analysis of individual tumors starts to realize the potential of post-genome era science and may finally deliver the ultimate goals of “individualized medicine” and “tailored therapy” – where treatment is fitted specifically to an individual.
The Best Treatment Is Prevention
Prevention requires a combination of activities involving different organizations, including public health strategies aimed at the whole population and exemplified by activities targeting adverse lifestyles, including smoking and poor diet. More targeted advice and possibly interventions may be needed for individuals at the highest predicted risk of disease. A new discipline of chemoprevention has been established with the sole purpose of designing the perfect weapon for a pre-emptive strike against future cancer cells. However, with the rare exceptions of individuals with known familial cancer syndromes, this has proved far more difficult a strategy for cancer prevention than it was for preventing coronary heart disease (CHD); there are no statin equivalents for cancer prevention.
At one extreme, no complex tests are needed to spot obese patients and smokers, and accurately predicting which of these will get early cancers as a result may be unnecessary, because encouraging all to change behavior appears a reasonable approach, particularly as in these cases such a lifestyle treatment is not likely to have any “off target toxicity” (prevent cancer but cause something as bad or worse). In other words, assuming that everybody is at risk of smoking and obesity-related disease may be good enough. Recent guidelines have placed vaccination of young women to prevent cancer-causing infection with HPV in this category. Being female is considered a sufficient risk of being infected with HPV and developing cervical cancer in the future and no subclassification is deemed desirable. Indeed, further selection could actually compromise the efficacy of vaccination as it might reduce the chance of developing herd immunity, although one could use sexual activity as an additional screening test and restrict vaccination to the noncelibate. In the case of invasive treatments, clearly it is preferable to narrow down the risk much more before offering preventative surgery or toxic drugs unless these are targeted correctly. Thus, before offering mastectomy to a woman to prevent breast cancer we need to know a lot more than just her gender. In this case the presence of very high-risk mutations in BRCA1 and BRCA2 identify a small subset of women who will benefit from surgery. However, there really is not much in between these two extremes. In other words, for most of us there are no simple tests that can be used to predict our risk of future cancer.
Robust tools have been developed allowing reasonably accurate estimation of future risk of heart attacks or strokes based on using simple information such as age, sex, blood pressure, and level of circulating fats (readily determined in the clinic) in order to calculate a risk score. For cancer the hope is that improved genetic testing (Box 1.3), measurement of new disease biomarkers, and improved clinical investigational tools will match these successes in time. Screening is discussed in more detail below and in Box 1.1.
What Is Next Best?
The early detection of cancer or precancer syndromes is self-evidently the next best to prevention, based on the assumption that small numbers of well-localized cells of a potentially less advanced state of malignancy will prove easier to treat or cure. This forms the basis of screening for cervical, breast, and colon cancers (Box 1.1). Improved early detection also involves the speedy selection of patients with appropriate symptoms or signs for early application of diagnostic tests (including X-rays, blood tests, biopsy, etc.). The nature of such tests is continually evolving, with great hope placed on the identification of cancer biomarkers and resultant possibility of molecular diagnostics gradually supplanting or complementing more traditional morphological assessments. Biomarkers may derive from a variety of sources, including serum proteins or nucleic acids, circulating cancer cells singly, or as part of complex molecular signatures. Not all such new diagnostic tests will necessarily result from ever more advanced molecular and cellular biology. Some of the ideas of the original pioneers of cancer biology still have potential and are being evaluated (see Jean Astruc, in Appendix 1.1), even highly creative or eccentric ideas such as training “sniffer dogs” to identify bladder cancer from the smell of a person’s urine (though with any dog I’ve met the trick appears to be to stop them publicly “screening” everybody within reach!).
Currently Available Treatment Options
The number of treatment options has expanded dramatically in recent years with the emergence of specific therapies targeting individual cancer-relevant molecules or signaling pathways. Thus, knowledge that a cancer is possessed by a particular malign oncogenic mutation can be exploited by the administration of a suitable therapeutic exorcism. However, choice of appropriate treatment regimens for any given patient remains challenging. In general, the first decision to be made is whether the cancer may be cured by surgical resection and radiation or drugs, or both. A more detailed discussion of cancer therapies is presented in Chapter 16, but a few interesting aspects will be highlighted here.
Achieving lasting remission in patients suffering from nonlocalized malignancies remains elusive. We are rarely, if ever, able to kill all the cancer cells in the primary tumor and metastatic lesions. Such failures may be the result of poor access of effective treatments to all tumor locations, varying susceptibility to conventional DNA-damaging anticancer agents, or the rapid evolution of resistance. A particular problem is posed by cancers where cells spread early via the circulation to establish micrometastases in the bone marrow or elsewhere. While increasing drug dosage can overcome some of these barriers it also increases toxicity to normal cells; to paraphrase Paracelsus, “The dose makes the poison.”
Traditional cytotoxic treatments aim to kill all cancer cells, whereas some newer approaches may be directed at disabling cancer cells (inducing growth arrest, differentiation, etc.), without necessarily killing them. Therapeutic resistance is a major issue in cancer treatments and may arise by cancer cells acquiring new routes of signaling that bypass the drug-targeted protein or even by developing ways of blocking the drugs access to the cell. Cancer stem cells are also a potential explanation for treatment resistance and recurrence, as such cells may be inherently more resistant to agents or may occupy environments such as hypoxic niches which protect them.
Despite some notable successes, concerns remain about potential adverse effects of traditional radio- and chemotherapy on normal tissues, and intriguingly also on the surviving cancer cells themselves. Cancer progression is an evolutionary process driven by acquisition of epimutations, which provide a selective growth advantage to particular cell populations. Therapies that induce irreparable damage to cell DNA may have undesirable consequences on cancer cells – they may fail to undergo apoptosis and go on to survive the onslaught. In fact, one mechanism of resistance in cancer cells may be the increased mutation rate and selection pressure provided by such drugs. The net effect of unsuccessful cancer therapies could be to speed the progress of the disease, as more mutated cells expand without the competition of their less aggressive predecessors and their offspring. We might actually help to select for more aggressive clones or enrichment of more malign cells such as CSCs. Increasingly, therefore, new combinations of drugs are employed to reduce the likelihood of cancer cells surviving to become resistant to all these agents.
An interesting parallel may be drawn here between evolution of species and evolution of a cancer. Evolution is driven not only by mutations and natural selection, but also by catastrophic extinctions which, by removing less-hardy competitors, clear the path for the survivors to fill the vacuum. It may be that subtotal cancer cell killing with chemotherapy, radiotherapy, or even surgery is the cancer equivalent of a meteor impact. Given, that only some 1 in 10 000 of the estimated 50 billion or more species that have evolved on earth still exist, and that if we are anything to go by the survivors include some of the hardiest and nastiest, then perhaps extinctions of some of the less able to survive may be undesirable if you do not cull the lot. Moreover, the situation in cancer therapy is likely a lot worse, as cancer cells are repeatedly selected for their ability to not be killed by cancer therapy, whereas species have not necessarily been selected largely on their ability to survive repeated meteor impacts or volcanic activity but probably somewhat more randomly. The risks inherent to increasing “selection pressure” have been ably demonstrated in the case of emergence of antibiotic resistance in bacteria.
So what might be an effective alternative to treatments involving chemo-, radiotherapy or surgery? Theoretically, arresting replication in cancer cells might be a good alternative or addition to traditional treatments that offer anything other than complete extinction of cancer cells, as this would prevent expansion of an aggressive surviving clone and might instead foster “stagnation” of the cancer cell population. Assuming that no treatment will ever immediately kill all cancer cells – what proportion effectively constitutes total extinction? The 90% extinction of species believed to have occurred in the Permian era was followed by a substantially slower recovery (based on fossil records and therefore really only applies accurately to “big organisms”) than after those in different eras which resulted in 60–70% extinction – but they still eventually recovered. Arguably, we might wish to know what proportion of cancer cells need to be killed in an individual for no symptomatic recurrence of the tumor to take place during that individual’s lifespan!
Causes of Cancer
Much has been learned about the causes of cancer, including the role of genetic predisposition, gene–environment interactions, and infectious agents. Intriguingly, recent research points to the considerable overlap between the behavior of cancer cells and that of cells during normal physiological wound healing and during embryogenesis. Similarities include replication, less differentiated state, invasion/migration, with the major differences reflecting the lack of control and the unscheduled nature of replication that characterize cancer. One intriguing question, addressed later, is how the organism is able to distinguish between normal growth and tissue repair (normal cell cycles) on the one hand and neoplastic growth (cancer cell cycles) on the other.
The Clonal Evolution Theory
Most cancers derive from an individual somatic cell in the adult organism, with the initiation and progression of tumorigenesis dependent on the accumulation of genetic or epigenetic changes that determine the emerging cancer phenotype. Initiation is believed to be through DNA damage, which renders the cell capable of forming a cancer; initiated precancer cells then multiply during a promotion phase. One way of looking at this is to assume that the first mutation in some way liberalizes the would-be cancer cell, thereby generating an underclass of uniquely susceptible cells among which some may subsequently become increasingly radicalized. Cancer cells are created by the assimilation of epimutations that promulgate increasingly individualistic and sociopathic behaviors at odds with the best interests of the organism and, moreover, this process may proceed through recognizable stages.
A “multistage” model of carcinogenesis, based largely on epidemiological observations, was first articulated by Peter Armitage and Richard Doll in the 1950s. The rapid expansion of knowledge about the molecular genetic basis of disease then allowed Nowell, in 1976, to suggest that cancers arise by a process of multistep clonal evolution. He proposed that most neoplasms arise from a single cell of origin, and tumor progression results from acquired genetic variability within the original clone, allowing sequential selection of more aggressive sublines. He also stated, rather prophetically, that acquired genetic instability may result in apparently similar individual advanced tumors being very heterogeneous both at a molecular and behavioral level and might require individual specific therapy. He also predicted that therapy could be thwarted by the emergence of genetically variant resistant sublines.
Becoming a Cancer Cell – Multistage Carcinogenesis
A wealth of data has supported the view that cancers are multistage diseases progressing via protracted accumulation of multiple genetic and/or epigenetic changes (lesions) that compromise control of cell proliferation, survival, differentiation, migration, and social interactions with neighboring cells and stroma.
Hanahan and Weinberg have recently updated their seminal review of 2000, in which they originally construed the axiomatic requirements of cancer cells as:
Not surprisingly, other cancer-critical processes have been vying for the dubious accolade of becoming the seventh hallmark feature of cancer ever since. The Warburg effect, a shift in energy production from oxidative phosphorylation to glycolysis, is currently leading the polls, but is being hard pressed by avoidance of immune surveillance, tissue remodeling, and a variety of forms of stress or stress phenotype. In most cancers the presence of chronic inflammation alongside subversion of expected interactions with immune and stromal cells also appear to be common features.
By implication, tumor progression proceeds by the acquisition of lesions that provide the tumor cell with these attributes and which thereby shape the complex phenotype of the tumor cell (Fig. 1.8). Mostly, these lesions are acquired in somatic cells, but in the inherited cancer syndromes (see Chapter 3), one of the lesions is inherited and is present in all somatic cells – the would-be cancer cell has a headstart in life. It is important to note that seemingly phenotypically similar cancers may arise through differing combinations of lesions: there are likely many different routes to cancer, even in the same cell type (Box 1.4). Many key cancer-relevant signaling pathways may be activated or inactivated by mutations at various different points that could result in largely identical cell behaviors. Many of the “hallmark” features of cancer cells may be the consequence of reactivating embryonic developmental programs by different routes and will be discussed later with respect to CSCs and EMT.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


