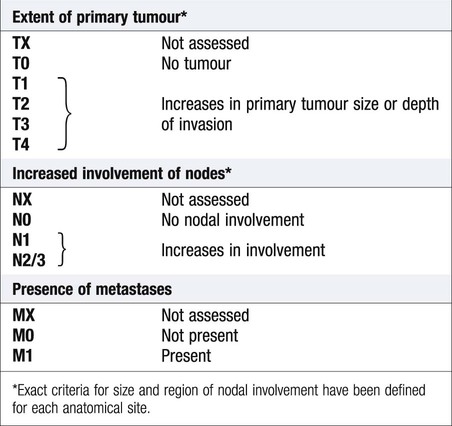
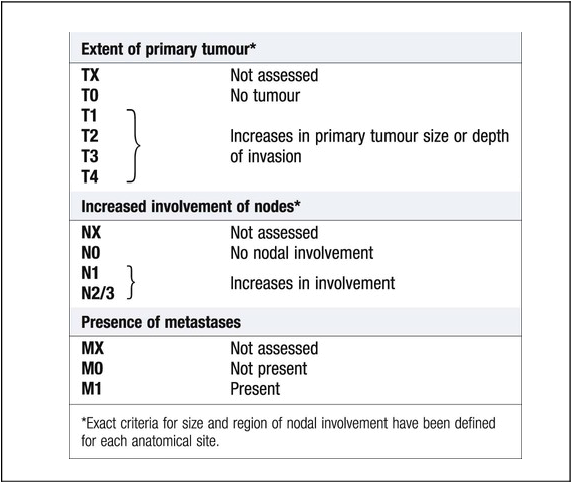
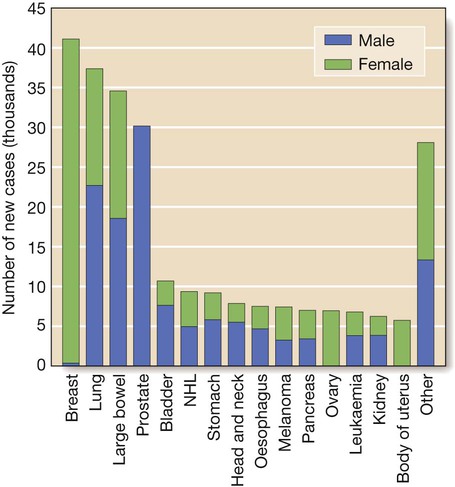
Cancer is a significant global health-care problem, with an estimated worldwide incidence of 10 million new cases per year, 46% of which are in developed countries. Mortality is high, with more than 7 million deaths per year. The global costs and socioeconomic impact are considerable. The most common solid organ malignancies arise in the lung, breast and gastrointestinal tract (Fig. 11.1), but the most common form worldwide is skin cancer. Tobacco is a major factor in the aetiology of 30% of cancers, including those of the lung, nasopharynx, bladder and kidney, and these could be prevented by smoking cessation. Diet and alcohol contribute to a further 30% of cancers, including those of the stomach, colon, oesophagus, breast and liver. Lifestyle modification could reduce these if steps were taken to avoid animal fat and red meat, reduce alcohol, increase fibre, fresh fruit and vegetable intake, and avoid obesity. Infections account for a further 15% of cancers, including those of the cervix, stomach, liver, nasopharynx and bladder, and some of these could be prevented by infection control and vaccination. There are three principal mechanisms through which cell death occurs in healthy tissues: • Apoptosis is programmed cell death and is frequently found at markedly reduced rates in cancers, particularly those of high grade or those resistant to treatment. The cellular apoptotic system has regulatory elements which sense intrinsic and extrinsic pro-apoptotic signals and initiate a cascade of proteolysis and cell disassembly with nuclear fragmentation, chromosomal condensation, and shrinking of the cell with loss of intercellular contact, followed by cellular fragmentation and the formation of apoptotic bodies that are phagocytosed by neighbouring cells. The most important regulator of apoptosis is the TP53 tumour suppressor gene, often described as the ‘guardian of the genome’, as it is able to induce apoptosis in response to sufficient levels of genomic damage. The largest initiator of apoptosis via TP53 is cellular injury, particularly due to DNA damage from chemotherapy, oxidative damage and ultraviolet (UV) radiation. • Autophagy is a catabolic process during which cellular constituents are degraded by lysosomal machinery within the cell. It is an important physiological mechanism, which usually occurs at low levels in cells but can be induced in response to environmental stresses, particularly radiotherapy and cytotoxic chemotherapy, which induce elevated levels of autophagy that are cytoprotective for malignant cells, thus impeding rather than perpetuating the killing actions of these stress situations. Severely stressed cancer cells have been shown to shrink via autophagy to a state of reversible dormancy. • Necrosis is the premature death of cells and is characterised by the release of cellular contents into the local tissue microenvironment, in marked contrast to apoptosis, where cells are disassembled in a step-by-step fashion and the resulting cellular fragments phagocytosed. Necrotic cell death results in the recruitment of inflammatory immune cells, promotion of angiogenesis, cellular proliferation and tissue invasion. Necrotic cells also release stimulatory factors, which promote proliferation of neighbouring cells and can promote rather than inhibit carcinogenesis. The cell cycle is comprised of four ordered, strictly regulated phases referred to as G1 (gap 1), S (DNA synthesis), G2 (gap 2) and M (mitosis) (Fig. 11.2). Normal cells grown in culture will stop proliferating and enter a quiescent state called G0 once they become confluent or are deprived of serum or growth factors. The first gap phase (G1) prior to the initiation of DNA synthesis represents the period of commitment that separates M and S phases as cells prepare for DNA duplication. Cells in G0 and G1 are receptive to growth signals, but once they have passed a restriction point, they are committed to enter DNA synthesis (S phase). Cells demonstrate arrest at different points in G1 in response to different inhibitory growth signals. Mitogenic signals promote progression through G1 to S phase, utilising phosphorylation of the retinoblastoma gene product (pRb). Following DNA synthesis, there is a second gap phase (G2) prior to mitosis (M), allowing cells to repair errors that have occurred during DNA replication and thus preventing propagation of these errors to daughter cells. Although the duration of individual phases may vary, depending on cell and tissue type, most adult cells are in a G0 state at any one time. All cancers require a functional vascular network to ensure continued growth and will be unable to grow beyond 1 mm3 without stimulating the development of a vascular supply. Tumours require sustenance in the form of nutrients and oxygen, as well as an ability to evacuate metabolic waste products and carbon dioxide. This entails the development of new blood vessels, which is termed angiogenesis (Figs 11.3 and 11.4). Invasion and metastasis are complex processes involving multiple discrete steps; it begins with local tissue invasion, followed by infiltration of nearby blood and lymphatic vessels by cancer cells. Malignant cells are eventually transported through haematogenous and lymphatic spread to distant sites within the body, where they form micrometastases that will eventually grow into macroscopic metastatic lesions (see Fig. 11.3). However, deficiencies in the development or function of CD8+ cytotoxic T lymphocytes, CD4+ Th1 helper T cells, or natural killer cells can each lead to a demonstrable increase in cancer incidence. Also, highly immunogenic cancer cells may evade immune destruction by disabling components of the immune system. This is done through recruitment of inflammatory cells, including regulatory T cells and myeloid-derived suppressor cells, both actively immunosuppressive against the actions of cytotoxic lymphocytes (see Fig. 4.6, p. 80). Environmental triggers for cancer have mainly been identified through epidemiological studies that examine patterns of distribution of cancers in patients in whom age, sex, presence of other illnesses, social class, geography and so on differ. Sometimes, these give strong pointers to the molecular or cellular causes of the disease, such as the association between aflatoxin production within contaminated food supplies and hepatocellular carcinomas. However, for many solid cancers, such as breast and colorectal, there is evidence of a multifactorial pathogenesis, even when there is a principal environmental cause (Box 11.1). A number of inherited cancer syndromes are recognised that account for 5–10% of all cancers (Box 11.2). Their molecular basis is discussed in Chapter 3, but in general they result from inherited mutations in genes that regulate cell growth, cell death and apoptosis. Examples include the BRCA1, BRCA2 and AT (ataxia telangiectasia) genes that cause breast and some other cancers, the FAP gene that causes bowel cancer, and the Rb gene that causes retinoblastoma. Although carriers of these gene mutations have a greatly elevated risk of cancer, none has 100% penetrance and additional modulating factors, both genetic and environmental, are likely to be operative. Exploration of a possible genetic contribution is a key part of cancer management, especially with regard to ascertaining the risk for an affected patient’s offspring. The overall fitness of a patient is often assessed by the Eastern Cooperative Oncology Group (ECOG) performance scale (Box 11.3). The outcome for patients with a performance status of 3 or 4 is worse in almost all malignancies than for those with a status of 0–2, and this has a strong influence on the approach to treatment in the individual patient. The process of staging determines the extent of the tumour; it entails clinical examination, imaging and in some cases surgery, to establish the extent of disease involvement. The outcome is recorded using a standard staging classification that allows comparisons to be made between different groups of patients. Therapeutic decisions and prognostic predictions can then be made using the evidence base for the disease. One of the most commonly used systems is the T (tumour), N (regional lymph nodes), M (metastatic sites) approach of the International Union against Cancer (UICC, Box 11.4). For some tumours, such as colon cancer, the Dukes system (p. 914) is used rather than the UICC classification. • Signet-ring cells favour a gastric primary. • Presence of melanin favours melanoma. • Mucin is common in gut/lung/breast/endometrial cancers, but particularly common in ovarian cancer and rare in renal cell or thyroid cancers. • Psammoma bodies are a feature of ovarian cancer (mucin +) and thyroid cancer (mucin −). • Oestrogen (ER) and progesterone (PR) receptors. Positive results indicate that the tumour may be sensitive to hormonal manipulation. • Alpha-fetoprotein (AFP) and human chorionic gonadotrophin (hCG) ± placental alkaline phosphatase (PLAP). These favour germ-cell tumours. • Prostate-specific antigen (PSA) and prostatic acid phosphatase (PAP). These favour prostate cancer. • Carcinoembryonic antigen (CEA), cytokeratin and epithelial membrane antigen (EMA). These favour carcinomas. • HER2 receptor. Breast cancers that have high levels of expression of HER2 indicate that the tumour may respond to trastuzumab (herceptin), an antibody directed against the HER2 receptor.
Oncology
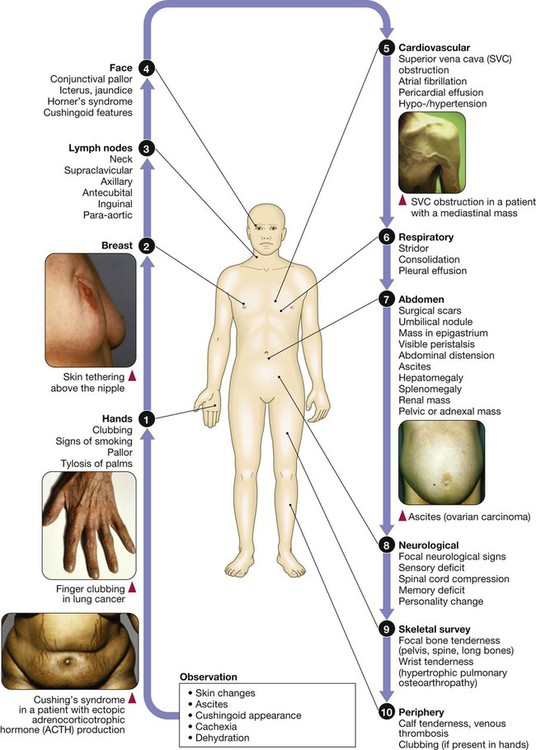
Clinical examination of the cancer patient
The ten hallmarks of cancer
2 Resisting cell death
3 Sustaining proliferative signalling
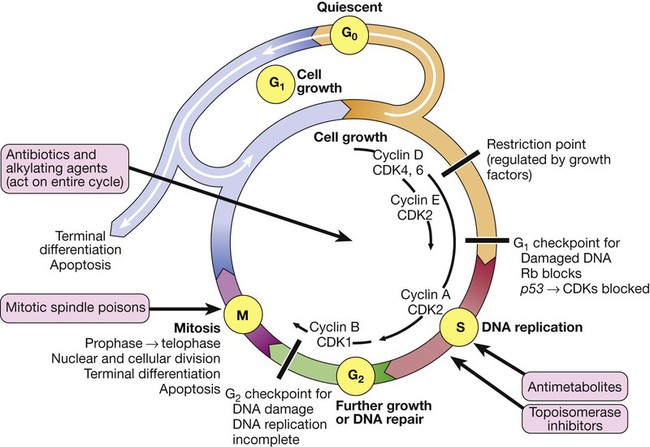
The cell cycle
6 Inducing angiogenesis
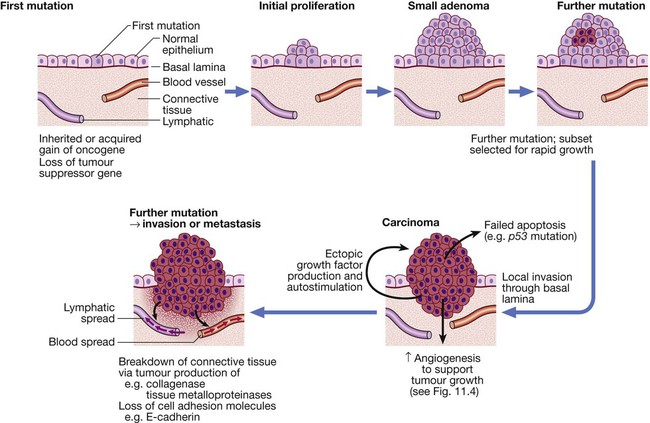
The multistep origin of cancer, showing events implicated in cancer initiation, progression, invasion and metastasis.
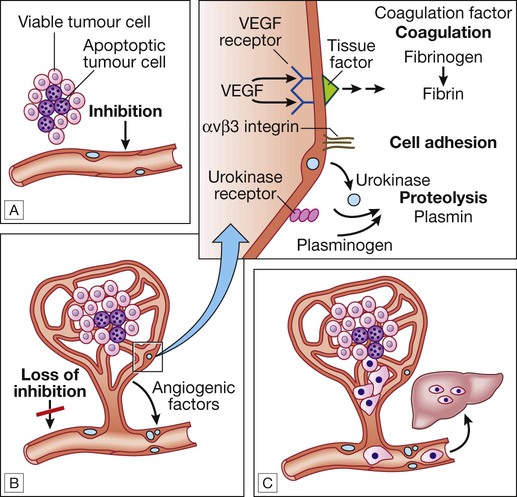
A For any cancer to grow beyond 1 mm3 it must evoke a blood supply. B New vessel formation results from the release of angiogenic factors by the tumour cells and loss of inhibition of the endothelial cells. C The loss of cellular adhesion and disruption of the extracellular matrix allow cells to extravasate into the blood stream and metastasise to distant sites.
7 Activating invasion and metastasis
10 Evading immune destruction
Environmental and genetic determinants of cancer
Environmental factors
Genetic factors
Investigations
Histology
Light microscopy
Immunohistochemistry
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Oncology
 Examination of the skin
Examination of the skin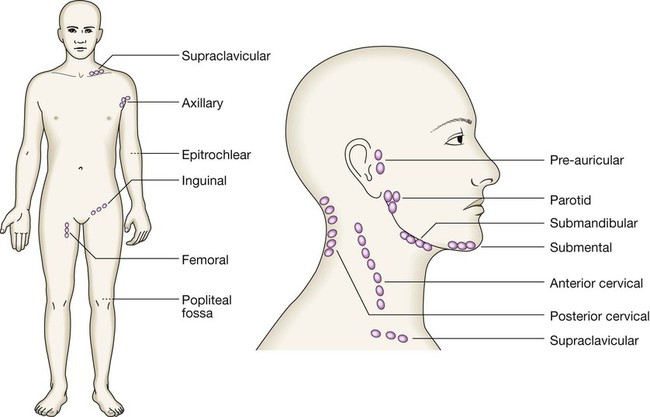
 11.1
11.1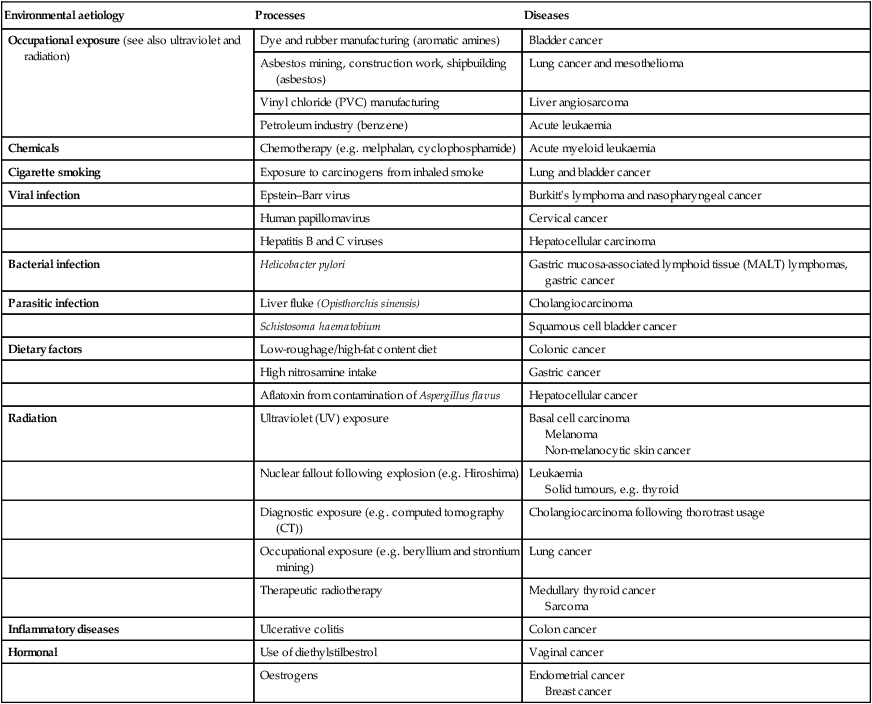
 11.2
11.2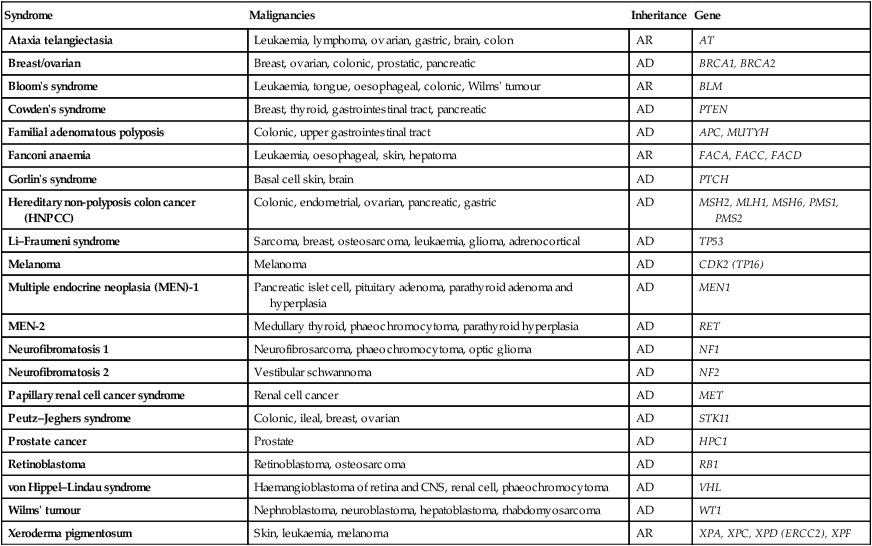
 11.3
11.3 11.4
11.4