- Oncogenes (cancer-causing genes) are essentially anarchic versions of normal cellular genes. Once their antisocial behavior is unleashed, as a result of mutations or other means, they either produce inappropriate amounts of protein or an altered protein that is fixed in the “on” position.
- The normal gene versions of oncogenes are referred to as proto-oncogenes and their products are proto-oncoproteins, giving the misleading impression that they are defined entirely by their potential to cause cancer. Care is needed lest we wrongly relegate the often pivotal roles played by proto-oncoproteins in normal cellular homeostasis to a footnote; this terminology is historical and references the route by which they were first recognized.
- The modern era of cancer biology was ushered in by the discovery of oncogenes and is heavily indebted to the insightful studies of sarcomas in chickens that turned out to be the result of an infectious agent (named the Rous sarcoma virus, after Peyton Rous, who identified it). The viral oncogene v-src was subsequently pinpointed as the key culprit and the story was nicely rounded off when Mike Bishop and Harold Varmus found the normal cellular counterparts of the viral oncogene, c-SRC.
- With the recognition that viruses were not required to deliver activated oncogenes to would-be cancer cells and that these could be generated in-house, it was not long before the processes by which this might occur were described. We now know that oncogenes become activated by genetic mutations, chromosomal translocations, or gene amplification. In some cases, such as HPV infection, viruses are still central players, but as we have seen in Chapter 3, mostly the DNA damage occurs in situ within the nucleus of the target cell.
- The proto-oncogenes encode proteins that direct such pivotal cellular functions as cell replication, growth, survival, loss of differentiation, and motility. Thus, by definition, the oncogenic variants will drive these processes independently of normal regulatory signaling pathways.
- In general, an activating mutation in a single copy of an oncogene is sufficient to evoke a tumorigenic process, whereas, in contrast, both gene copies of a tumor suppressor must usually be inactivated in order for the restraints on that process to be removed.
- It is also important to note that oncogenes, as is ironically often the case for c-SRC and also c-MYC, may become upregulated (activated) and help drive tumorigenesis without mutations in either the coding or regulatory regions of their own genes. Thus, levels of expressed proteins can be inappropriately increased by epigenetic factors such as loss of imprinting (see Chapter 11), increased stability of the oncoprotein (due to alterations in processing pathways), or by deregulated upstream signaling, which will not be revealed by looking for changes in the coding sequence (mutations) in that oncogene.
- In fact, the relative paucity of mutations in c-MYC and c-SRC resulted in gross underestimation of what we now know are the central roles played by these oncogenes in the evolution and maintenance of arguably most human cancers.
- Following the structure used in the previous chapter, we will consider oncogenes under five familiar functional headings: growth factors, receptors, signal transducers, transcription factors, and those that primarily act on prevention of the mitochondrial (intrinsic) pathway of cell death – apoptosis (e.g. BCL-2 and BCL-XL).
- Individual oncogenes do not cause cancer on their own but rather act in concert with other oncogenes or loss of tumor suppressors – termed “oncogene collaboration” or “oncogene cooperation.” Propitious epimutations that provide a growth advantage by complementing those acquired previously will be selected for and the affected cell will give rise to a new clone that will usurp its predecessors.
- Oncogene cooperation has been intensively studied in cell lines and using various models of cancer in vivo. More recently, the generation of regulatable expression systems has allowed the role of various oncogenes in tumor maintenance to be explored and “proof of hypothesis” studies for oncogene-targeted therapies to be conducted before attempting the complex task of designing and testing new small molecule drugs (see Chapters 15 and 16).
- Recent work has indicated the importance of differing levels of oncogene expression or activation on phenotype and thereby also on tumor evolution. Paradoxically, high levels of expression and activation of oncogenes, such as c-MYC and RAS, can actually stall tumor progression by engaging their own “tumor suppressor” properties to promote cell death or senescence, respectively, whereas low levels can drive oncogenesis without engaging tumor suppression.
- Taken together, this creates the novel counterintuitive possibility of treating cancers by actually “overdriving” oncogenic pathways as an alternative to antagonizing them. This approach might prove more tractable than trying to restore a malfunctioning tumor suppressor pathway that has by definition failed in its duty to restrain aberrant RAS or MYC activity. However, there is a critical caveat – namely, that the “intensified” oncogenic signal is still capable of engaging a functioning death/senescence pathway.
Introduction
Turning and turning in the widening gyre. The falcon cannot hear the falconer; Things fall apart; the centre cannot hold; Mere anarchy is loosed upon the world, The blood-dimmed tide is loosed, and everywhere the ceremony of innocence is drowned.
William Butler Yeats
Having digested the previous chapter, you will now be aware that many of the genes impinging on signaling pathways are designated as oncogenes or tumor suppressors, depending on whether they promote or inhibit growth. This chapter and the following one will discuss these central regulators in more detail and illustrate key concepts through a very detailed discussion of specific examples, singled out because of their particular importance or because these genes/proteins have wide-ranging functions beyond those of traditional views of signaling molecules.
As has been discussed in Chapters 4 and 5, somatic cells only enter the cell cycle on demand and then under the strict control of mitogenic signals delivered in the appropriate way. These signals, usually in the form of secreted growth factors, bind to cell surface receptors and trigger the cascades of intracellular signals that enable cells to progress through the G1/S transition. Growth-promoting pathways are tightly regulated at multiple sites along the cascade, creating complex signaling networks that ensure cells proliferate only when this is useful to the whole organism, including after tissue injury or to replace normal cell losses and not if the DNA is damaged.
Loss of these normal restraints can result in anarchy, as cells and their offspring increasingly dance to their own tune with scant regard for the needs of the organism. Mutations in key regulatory genes can disrupt signaling networks, resulting in unscheduled cell division, growth and avoidance of cell death, in defiance of normal controls – setting the scene for cancer. At the present time over 450 genes are known to contribute to the development of cancer in humans. Although the specific genes affected and the order in which mutations occur differ from one cancer patient to another (the “cancer road-map”), the consequent effects of these epimutations on cancer cells impinge on a finite number of common key regulatory pathways. In fact, diversity at a molecular level does not necessarily equate with similar profligacy at the level of cell behavior, as exemplified by the limited repertoire of shared phenotypic hallmark features of cancer cells described earlier. This notwithstanding, a precise description of the molecular pathway to oncogenesis is still critically important and in notable cases has already led to the identification of prognostic biomarkers and new treatment targets that may not have been revealed in any other way.
The identification and characterization of the genes involved in cancer – the oncogenes, tumor suppressors, and most recently caretaker genes, has been one of the great triumphs of molecular biology. This concept has already been discussed in general terms in Chapter 3. This chapter will concentrate on the oncogenes, Chapter 7 will discuss the tumor suppressors, and Chapter 10 the caretaker genes.
The Oncogenes
Oncogenes (from Greek onkos, a tumor) are activated versions of normal genes (proto-oncogenes) that generally share the ability to accelerate cell division and growth, but often also variously contribute to loss of differentiation, increased cell motility, and avoidance of apoptosis and invasion. (Note: The nomenclature whereby a normal gene involved in important cellular functions relating to growth is defined by the role it might play in a cancer if released from normal restraints may seem at first glance a peculiar one. However, this is a historical legacy reflecting the way in which they were first discovered.)
In healthy cells, the level of expression and activity of the proto-oncogenes are tightly regulated. This is essential as these are powerful proteins with the potential to provoke excessive cell proliferation or even the inappropriate survival of a cell with DNA damage. Oncogenes are distinguished from their normal cellular counterparts because they are overexpressed or because mutations result in the formation of protein products that are abnormally active or long-lived. In this scenario an abnormality in just one of the two gene copies in a cell is sufficient to generate the aberrant phenotype and the mutation is considered “dominant.” Conversely, and because most signaling pathways have both positive and negative regulators, the same net effect can be obtained by mutations which inactivate both copies of a tumor suppressor gene, thereby releasing the restraint normally imposed by the encoded tumor suppressor protein. In contrast to oncogenes, both copies of the gene must be lost or inactivated to bring about these effects, and so the mutant phenotype is considered “recessive.” However, as nothing is ever simple, it must be recognized that tumor suppressors may be “haploinsufficient” (can contribute to tumorigenesis when only a single copy is inactivated and the “dose” of tumor suppressor protein reduced by around 50%). At the risk of being repetitive, identical cellular behaviors can result from inactivation of tumor suppressors as from activation of oncogenes – most of the control mechanisms in the cell involve both inhibitory (tumor suppressor) and stimulatory (proto-oncogene) components. Put simply, the behavior of a cancer cell will reflect which regulatory or signaling pathways have become derailed rather than the means by which this has been achieved.
Moreover, to complicate matters further, intriguing studies over the last 20 years have forced a major reassessment of traditional views of the role of oncogenes in cancer. Thus, despite their undisputed importance in tumorigenesis, it is now clear that oncogenes (such as c-MYC, E2F, and RAS) capable of driving uncontrolled cell-cycle progression also possess intrinsic tumor suppressive mechanisms that can trigger cell death (apoptosis) or cellular senescence – even if these may ultimately operate through traditional tumor suppressors such as RB, P53, and DNA damage responses. In other words, oncogenes may act as their own tumor suppressors! The ability of oncogenes to promote such contrasting cell fates in a given cell – proliferation and death – may at first seem somewhat contradictory and confusing to the reader. In fact, you may take comfort in the knowledge that the discovery of this phenomenon in the early 1990s (see Chapter 8) was no less of a surprise to the scientific community, but has since had an enormous impact on the way we now perceive the evolution as well as the future treatment of cancer. To this end, we will discuss the importance of such intrinsic tumor suppression later in this chapter (and in more detail in Chapters 8, 9, and 10), and how putative cancer cells have found ways to block tumor-suppressing activities of oncogenes, for example through oncogene collaboration. With this in mind we will also discuss members of the BCL-2 family of proteins that specifically act as oncogenes by suppressing apoptosis.
One final point that may prove among the most important discoveries of recent years is that the actual level of oncogene expression and activity has profound implications for tumor development and treatments. Briefly, while high levels of MYC and RAS promote apoptosis and senescence, respectively, low levels can actually drive oncogenesis without engaging tumor suppression. This fascinating area will be discussed later. These new insights into oncogene biology have also begun to impact on the development of new targeted therapeutics and there is thus considerable overlap between this chapter and Chapter 16.
Oncogenes include a diverse range of genes encoding proteins involved in the regulation of cell division, differentiation, death/survival, and cell motility (see Tables 6.2–6.5) and some key examples of representative oncogene protein products are shown in Fig. 6.1. Following a general overview, a select sample of key proto-oncogenes will be described in detail to illustrate the role of their protein products in normal tissue growth and how their normally tight regulation by mitogenic or survival signaling may be circumvented in oncogenesis. The previous chapters have described the basic cell-cycle machinery (see Chapter 4) and how cell-cycle entry and progression are regulated under the influence of mitogens and survival signals (see Chapter 5). Chapter 5 has also described how mutations in various signaling pathways, particularly in the tyrosine kinases, may contribute to tumorigenesis and as such overlaps with this one in many respects, not least because this chapter also describes how growth-regulating processes become derailed by the activation of oncogenes, by mutations or otherwise, in cancer. However, here we concentrate on the concept and discovery of oncogenes and will focus on two key exemplars, MYC and RAS, as well as others, which merit a more detailed discussion because of their central role in the history of cancer biology or because they involve more than just growth factor signaling pathways.
Table 6.1 Viral and cellular oncogenes
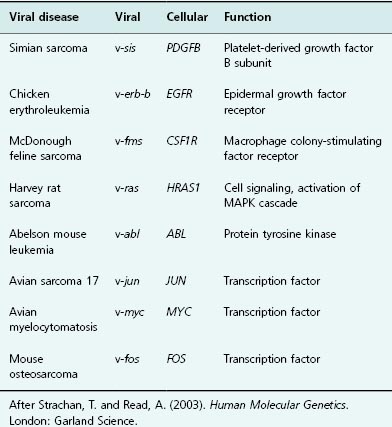
Table 6.2 List of oncogenes activated by amplification in cancer
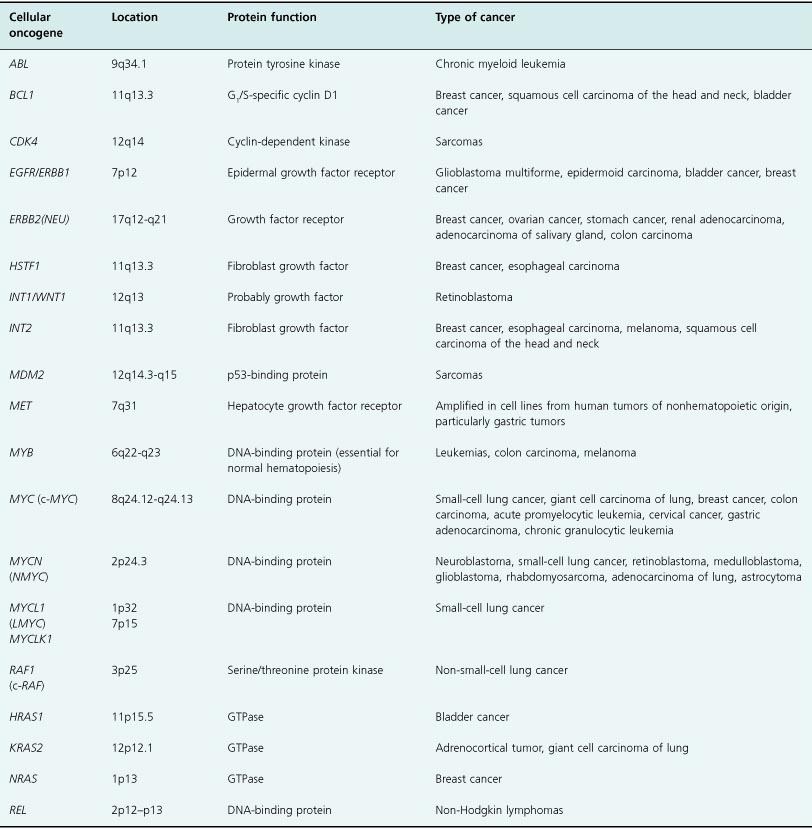
Table 6.3 Some known oncogenes classified by site of action in growth-regulating pathways
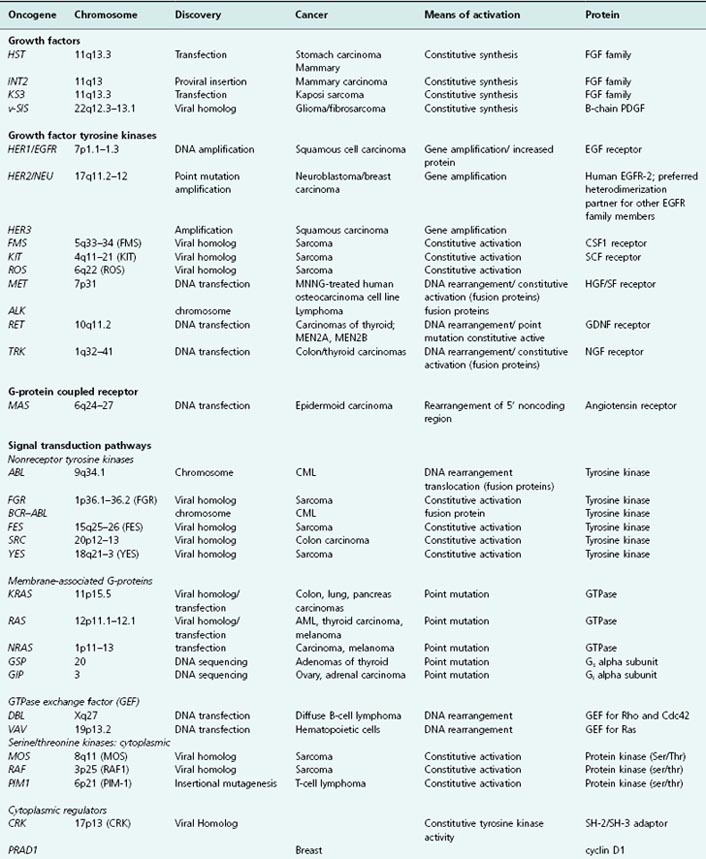
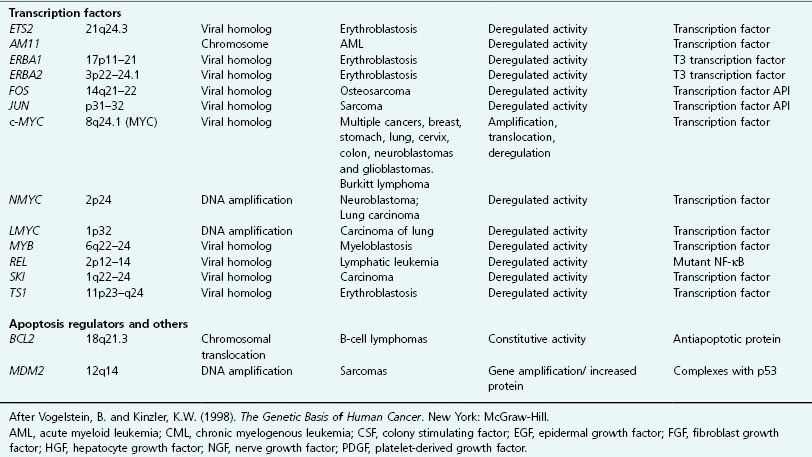
Table 6.4 Growth factor receptors and cancer
| Growth factor receptor | Activation | Cancer |
| ALK | Gene rearrangement: NPM–ALK t(2;5) | Anaplastic large-cell lymphomas, NSCLC |
| CSF-1R/FMS | Extracellular domain mutations | AML |
| CSFR/KIT | Point mutations and deletions | AML, mastocytomas, gastrointestinal stromal tumors |
| EGFR | Autocrine activation, amplification/overexpression | Squamous cell carcinoma, glioblastoma Colorectal cancer, NSCLC |
| EGFR | Extracellular domain deletions | Glioblastoma |
| HER2/NEU | Point mutation, amplification/overexpression | Breast and ovarian carcinoma |
| FGFR1 | Amplification | Breast cancer |
| FGFR1 | Chromosomal translocations | Myeloid malignancies |
| FGFR2 | Point mutations | Endometrial cancer |
| FGFR2 | Amplification | Gastric cancer |
| FGFR3 | Translocation | Multiple myelomas |
| FGFR3 | Point mutations | Bladder cancer, prostate |
| FLK2/FLT3 | Internal tandem duplication | AML, PML |
| C-MET | Gene rearrangement: TPR–MET t(1;7) | Stomach |
| C-MET | Amplification/overexpression | Thyroid, ovarian and colorectal cancers Acquired resistance in NSCLC |
| C-MET | Point mutations | Hereditary and sporadic papillary renal carcinoma |
| PDGFR | Autocrine activation | Osteosarcoma, melanoma, glioblastoma |
| PDGFR | TEL–PDGFRB t(5;12) translocation | CML |
| RET | Germline point mutations | Men2A, Men2B |
| TIE1, TIE2 | Paracrine activation | Tumor angiogenesis and lymphangiogenesis |
| TRKA | Gene rearrangement: TPM–TRKA t(1;1) | Papillary thyroid carcinoma |
| TRKC | Gene rearrangement: TEL–TRKC t(12;15) | Congenital fibrosarcoma, AML, breast carcinoma |
| VEGFR1, -2, -3 | Paracrine activation | Tumor angiogenesis and lymphangiogenesis |
AML, acute myeloid leukemia; CML, chronic myelogenous leukemia; PML, promyelocytic leukemia; NSCLC, non-small-cell lung carcinoma.
Table 6.5 Oncogenes in signaling pathways
| Signaling molecule | Activation | Cancer |
| H-RAS | Point mutations | Thyroid and bladder carcinoma |
| K-RAS | Point mutations | Pancreatic, lung and colon adenocarcinomas, non-small-cell lung carcinoma, myeloid leukemia, thyroid carcinomas |
| N-RAS | Point mutations | Melanoma, myeloid leukemia, thyroid carcinomas |
| B-RAF | Point mutations | Melanoma, colorectal carcinoma, small-cell lung cancer |
| SRC | Overexpression | Gastrointestinal cancers, lung, breast, ovary |
| BCR–ABL | Translocation | Chronic myelogenous leukemia |
| CYCLIN D1 | Amplification/overexpression | Breast cancer, head and neck squamous carcinoma, esophageal cancer, lymphoma |
| PI3K P110 | Amplification/overexpression | Ovarian and cervical cancer |
| STAT3 | Melanoma |
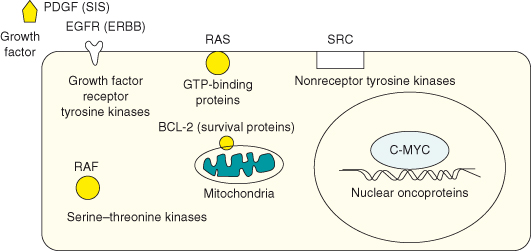
Figure 6.1 Representative oncogene products. In healthy cells, the normal nonmutated counterpart, proto-oncogenes, encode proteins that in general promote cell proliferation or cell survival but which are under tight regulation to avoid excessive proliferation, or indeed the survival of a cell that has sustained DNA damage and would normally be got rid of. Oncogenes include a diverse range of genes encoding proteins involved in the regulation of cell division, differentiation, and death or survival. The majority of oncogenes fall into five functional classes: transcription factors, growth factors, receptors, signal transducers, and survival proteins. Some key examples of representative oncogene protein products are shown here.
The Discovery of Oncogenes Ushers in the New Era of the Molecular Biology of Cancer
I pictured myself as a virus or a cancer cell and tried to sense what it would be like.
Jonas Salk
The concept of an oncogene (cancer-causing gene) was established by the observation of viral genes that could promote cancers in animal cells. The analysis of these animal viruses was directly responsible for the subsequent identification of genes associated with human cancer. Although it still holds true that most human cancers are not the direct result of viral infection, as had once been mooted, the similarity between the molecular cancer-causing genes in viruses and endogenous cellular genes involved in growth regulation and mutated in cancer gave a tremendous headstart to cancer molecular biology. In fact, in this way the identification of virus-related cancers in other animal species has had an immense impact on the development of modern theories of oncogenesis. It is worth reminding ourselves that, although subsequently relegated to the sidelines, the contribution made by viruses to human cancers has recently seen a renaissance (see Chapter 3). The discovery of oncogenes was not only a landmark event in unraveling the molecular and genetic basis of cancer, but was a major factor in furthering knowledge concerning the regulation of normal cell proliferation. In fact, one could claim that the foundations for the modern view of cancer formation were laid by chickens!
It had been suspected for a hundred years or more that viruses could cause cancer, but the first cancer-causing elements were described in viruses infecting poultry in 1909. Early studies by Peyton Rous on neoplasia in fowl demonstrated that retroviruses could induce tumors, but the relevance of these discoveries for the understanding of human cancer was disputed. Later, it was shown that the injection of these viruses was sufficient for tumor formation. As has been true of scientific progress throughout recorded history, the development of new tools preceded the development of new understanding. In this case, the availability of DNA recombinant techniques provided the means of studying cancer-causing elements in viruses and thence their cellular homologs. Arguably, this discovery in 1976, can lay claim to being the defining moment after which the molecular biology of cancer achieved primacy. When the viral DNA was examined, regions bearing strong similarity to genes that already existed in the animal were identified, suggesting that viruses had at some point incorporated host genomic sequences into their own genomes. By incorporating genes able to promote cellular growth and mutating them, the virus could enslave its host cell, increasing proliferation, and consequently, its own replicative potential. A glance at Table 6.1 will quickly reveal just how many oncogenes were first discovered from their viral homologs.
As it was rapidly appreciated that most common cancers did not arise through viral infection, some scientists predicted that, by extension, mutant cancer-causing genes must lie within all types of tumor cells, even those that lacked evidence of tumor virus infections. The availability of techniques for gene transfection led to the identification of cellular transforming genes and even some that at the time had no known viral counterpart. As predicted, tumor cells were found to contain such genes and when these mutated genes were introduced into normal fibroblasts they were sufficient to cause cancer. This gave rise to the idea that cells contain “proto-oncogenes” – genes that are concerned with normal aspects of cell behavior. The proto-oncogenes might become mutated and thus convert to “oncogenes” – genes that have the ability to convert normal cells to a cancerous state. The key studies that led to the discovery of oncogenes are described in Box 6.1.
Overview of Oncogenes
Man’s yesterday may ne’er be like his morrow; Nought may endure but Mutability.
Percy Bysshe Shelley
In this section, we discuss various oncogenes that were identified initially as viral oncogenes. Some well-known viral and cellular oncogenes are listed in Table 6.1. As mentioned above, oncogenes are mutant genes that have a dominant effect on cell growth; enhanced cellular replication and avoidance of cell death are common examples, and because of their central importance these processes are discussed further in Chapters 4 and 8, respectively. Historically, oncogenes were defined using cell culture experiments as genes able to “transform” normal cells into tumorigenic ones. Such transformed cells would manifest various characteristic abnormal cell behaviors in the culture dish, or be able to form tumors if implanted into a suitably immunosuppressed mouse host. Some of the common abnormal features of transformed cells include:
- uncontrolled cell proliferation in culture medium devoid of or depleted in serum or growth factors and
- the “piling up” of cells on top of each other to form characteristic multilayered foci that provide an experimentally convenient method of identifying transformed cells from their normal counterparts, which due to a strict requirement for attachment to a solid substrate only form a single layer of cells.
However, it is worth remembering that cell behavior in a culture dish – devoid of any stroma, removed from normal contacts with neighboring cells and extracellular matrix (see Chapter 12), as well as lacking signals derived from a vascular and lymphatic supply (see Chapter 14) – may differ from that of a cell in the intact organism, and the ramifications of this will be highlighted later in the chapter. Box 6.2, later in the chapter, introduces the nomenclature used for oncogenes.
Mechanisms of Converting Proto-Oncogenes to Oncogenes
The worst enemy of life, freedom and the common decencies is total anarchy; their second worst enemy is total efficiency.
Aldous Huxley.
Much of this has been discussed in Chapter 3, but a brief reminder will be included now. A proto-oncogene can be converted to an oncogene in a number of ways. In general, such mutations either affect the coding region of the gene, resulting in the formation of an abnormal oncoprotein with enhanced stability or activity, or may affect regulatory elements, resulting in enhanced or deregulated expression of the protein (Fig. 6.2). Oncogene mutations span a spectrum of genetic alterations, from simple point mutations altering only single amino acids in the protein product, to major chromosomal rearrangements that completely alter gene regulation (see Box 3.3). The major mechanisms involved in activating oncogenes can be classified as: structural alterations, from point mutations in a single gene to chromosomal translocations, or gene amplification, leading to overexpression of the oncogene.
Figure 6.2 Conversion of proto-oncogenes to oncogenes. A proto-oncogene can be converted to an oncogene in a number of ways. These include point mutations in a single gene (e.g. RAS) that can either affect the coding region of the gene resulting in the formation of an abnormal oncoprotein with enhanced stability or activity, or may affect regulatory elements resulting in enhanced or deregulated expression. Gene amplification can lead to overexpression of the oncogene. Chromosomal translocations or rearrangements can lead to overexpression of an oncoprotein, for example, in Burkitt’s lymphoma, the proto-oncogene c-MYC on chromosome 8 is translocated to one of the three chromosomes containing the genes that encode antibody molecules: immunoglobulin heavy chain locus (chromosome 14) or one of the light chain loci (chromosome 2 or 22). c-MYC now finds itself in a region of vigorous gene transcription leading to overproduction of the MYC protein. Lastly, the fusion of one protein to another might lead to its constitutive activity. For example, fusion of the promyelocytic leukemia (PML) protein to the retinoic acid receptor-alpha (RARα) generates the transforming protein of acute promyelocytic leukemias.
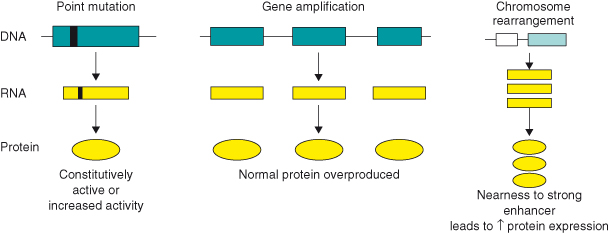
Table 6.2 shows some well-known examples of oncogenes activated by amplification in different cancers. Again, it is important to emphasize that oncoproteins may become overexpressed and contribute significantly to tumorigenesis by other means not involving gene mutation, such as via activation of various upstream signaling pathways.
Types of Oncogenes
If we had a keen vision of all that is ordinary in human life, it would be like hearing the grass grow or the squirrel’s heart beat, and we should die of that roar which is the other side of silence.
George Eliot
In this section we will give a brief introduction to a selection of well-known oncogenes. These oncogenes all play an important role in various cancers and in their treatments, some have been discussed in Chapters 4 and 5, and here we have selected a small number of oncogenes, c-MYC, RAS, and SRC, to discuss in more detail, because they are present in a large number of human cancers and give important historical and mechanistic insights into oncogenesis in general. A fairly comprehensive list of known oncogenes is given in Table 6.3, and what immediately becomes obvious is that the vast majority of these are involved in pathways that normally serve to regulate cell division and survival.
A recent study has examined a range of different human cancers for oncogenic mutations and the following 14 were most frequently observed: KRAS, NRAS, HRAS, BRAF, PIK3C, JAK2, EGFR, ERBB2, FGFR1, FGFR3, CDK4, RET, PDGFRA, and KIT (note the absence of both c-MYC and c-SRC from this list – these are upregulated/activated in most human cancers and play key roles in tumorigenesis, but rarely have mutations in either coding or noncoding regions).
A Simple Classification of Oncogenes
To retain respect for sausages and laws, one must not watch them in the making.
Otto von Bismarck
The vast majority of oncogenes are involved in pathways that normally serve to regulate cell division and survival (and other processes including cell motility and invasiveness – though it is likely that mutations influencing these behaviors would also need to confer some growth advantage in order to be selected for during cancer evolution). One way of subdividing oncogenes is, therefore, through the site in growth-regulating signal transduction pathways at which they act (we have deliberately tried to follow a similar classification to that used in Chapter 5). Thus, oncogenes may be consigned to five categories based on the functional and biochemical properties of their normal proto-oncogene/proto-oncoprotein counterparts. These are (1) growth factors, (2) receptors for growth factors and others, (3) signal transducers, (4) transcription factors, and (5) regulators of survival and others.
Growth Factors
The reader is referred to Chapter 5, where this subject is covered in more depth. Oncogenic activation of growth factors invariably results from transcriptional activation of the gene, leading to overproduction of the growth factor. The best-known growth factor oncogene is SIS, the oncogene incorporated into the genome of the simian sarcoma virus, and it encodes the platelet-derived growth factor PDGF B chain.
Receptors for Growth Factors and Others
Receptor Tyrosine Kinases (RTK)
Several mutations resulting in constitutively active receptors have been identified. For example, a single-point mutation converts the normal HER2 receptor into the NEU oncoprotein. Other RTK oncogenes have been identified and are listed in Table 6.4. The reader is again referred to Chapter 5 for a more detailed discussion.
G-Protein Coupled Receptors
There are few known examples of G-protein coupled receptors (see Chapter 5) acting as oncogenes, the most notable being the MAS gene, first identified in a mammary carcinoma. MAS is also believed to be involved in human epidermoid tumors and has been shown to encode an angiotensin receptor.
Signal Transducers
Nonreceptor Membrane-Associated Tyrosine Kinases
Members of this group include SRC, ABL, FGR (see Table 6.3) which can become oncogenic following DNA rearrangements, such as gene amplification or translocation as well as genetic mutations (Tables 6.2, 6.3, and 6.4). Whichever the event, the outcome is excessive tyrosine kinase activity in affected cells, often giving rise to various human leukemias.
The v-Src gene is historically of great importance and was the first oncogene to be identified (see Box 6.1). Moreover, the normal cellular gene c-SRC (SRC) is the archetypal protein tyrosine kinase (see Fig. 6.18), and the functional domains of other subsequently identified tyrosine kinases are still referred to on the basis of homology to those in SRC (SH-domains for SRC homology). The role of oncogenic SRC in cancer has experienced something of a renaissance in recent years and is discussed in detail later.
The prototypic nonreceptor tyrosine kinase c-ABL is implicated in various cellular processes but can become oncogenic following amplification of the gene, giving rise to chronic myelogenous leukemia (CML) (see Table 6.2). In addition, c-ABL can become oncogenic as a result of translocation associated with the Philadelphia chromosome, to form the BCR–ABL fusion protein, which is discussed later under its own heading as well as in the section on RHO–GEF. Importantly, this translocation event is found in over 90% of CMLs.
The RAS Family
Among the first human oncogenes to be identified and frequently mutated in cancers, RAS genes encode members of the RAS family of membrane-associated small G-proteins. Given their importance in human cancer, we will specifically discuss RAS and related family members in detail later in the chapter. There are three different oncogenic homologs of the c-RAS gene (see Table 6.5), each of which was identified in a different type of tumor cell. In all cases, oncogenic RAS genes have undergone point mutations that eliminate the intrinsic GTPase activity of the RAS protein. The result is that RAS is permanently “switched on.”
Serine/Threonine Kinases
The RAF gene was originally identified as a transforming oncogene from the rat fibrosarcoma virus, from which its name derives. Incorporation of the RAF gene into the viral genome resulted in truncation of the protein, with the resulting protein possessing constitutive kinase activity. Since the normal RAF gene product, RAF, is responsible for threonine phosphorylation of MAP kinase (MAPK) following receptor activation, oncogenic RAF leads to constitutive activation of the downstream MAPK pathway (see Fig. 6.14). The BRAF gene has been found to be mutated in some human cancers. Table 6.5 summarizes some of the known oncogenes in signaling pathways.
Transcription Factors
A considerable number of transcription factors have been shown to possess oncogenic activity, when deregulated. Not surprisingly, given the potentially large number of genes whose expression could be deregulated as a consequence. Examples of well-known oncogenic transcription factors include FOS, JUN, MYC, NFkB, GLI, MYB, ETS, and others (see Tables 6.1 and 6.2).
As discussed in Chapter 3, various mechanisms of oncogene activation have been demonstrated and depend on the host and the oncogene in question. Thus, oncogenes may be activated by integration of a virus in the vicinity of the host gene or through direct incorporation of the oncogene into a transforming retroviral genome. Oncogenes may also be activated by point mutations and by chromosomal translocation (see Box 3.3). In some cases, there is evidence for mutations that lead to an increased stability of the oncoprotein (e.g. c-MYC and FOS).
The v-fos gene was identified as a transforming oncogene of the feline osteosarcoma virus. The product of the human homolog gene, FOS, interacts with a second proto-oncoprotein, JUN, to form a transcriptional regulatory complex. Aberrant oncogenic activation of v-fos results from truncations of the coding and noncoding sequences of the gene following incorporation into the viral genome. This results in the loss of “destability” sequences as well as sequences involved in transcriptional repression of Fos. As a consequence, a stable FOS protein is produced and, unlike the normal situation in replicating cells where expression is transient, high levels of FOS may persist throughout the cell cycle.
The ETS family (E-twenty-six) is one of the largest families of transcription factors, characterized by a conserved ETS DNA-binding domain. Under normal regulation, ETS family members are involved in a variety of cellular functions including cell cycle, differentiation, migration, apoptosis, and angiogenesis and are downstream nuclear targets of RAS–MAP kinase signaling (see Fig. 6.13). The deregulation of ETS genes results in the malignant transformation of cells, including invasion, metastasis, and neo-angiogenesis. Several ETS genes are rearranged in human leukemia (e.g. TEL ETS gene fused to JAK2) and Ewing’s sarcoma (e.g. ERG ETS gene fused to EWS gene) to produce chimeric oncoproteins (fusion of elements of at least two different proteins).
The c-MYC gene was originally identified in the avian myelocytomatosis virus. The oncogenic functions of the transcription factor c-MYC play a key role in well-known human cancers, such as Burkitt’s lymphoma as a result of chromosomal translocation (see Chapter 3, Box 3.3). In addition, this oncogene appears to be activated in the majority of human cancers at some stage during tumor development. For this reason, we have selected c-MYC for further discussion in the next section, and will highlight both its normal biological functions as well as those during oncogenic activation in vitro and in genetically altered mouse models. Although the mechanism for c-MYC activation remains unclear in many human cancers, there is evidence of mutations that confer increased stability of the c-MYC protein. In all cases though, the transforming effect of oncogenic c-MYC (described in the next section) is the result of its elevated or deregulated levels of expression.
Regulators of Cell Survival and Death
Normal tissues are maintained by a regulated balance between cell proliferation and cell death (apoptosis). In fact, apoptosis is a normal process during embryonic development and tissue homeostasis in the adult – otherwise referred to as programmed cell death (Chapter 8 covers this in depth). Studies of cancer cells have shown that both uncontrolled cell proliferation and failure to undergo apoptosis can contribute to neoplasia and insensitivity to anticancer treatments. Many proto-oncogenes may affect survival in some way, usually indirectly. Of those that specifically regulate apoptosis, members of the BCL2 family (see Chapter 8), first identified by the study of chromosomal translocations in human lymphoma (see Box 3.3) are the most studied to date.
The BCL2 gene encodes a protein, BCL-2, localized to the inner mitochondrial membrane, endoplasmic reticulum, and nuclear membrane. This BCL-2 family is now known to include a large number of proteins with either anti- or proapoptotic activities. If expression of antiapoptotic BCL-2 family members is increased, or that of proapoptotic members lost, then cells are vulnerable to other cancer-causing mutations, in particular those which increase proliferation. Such cells will now no longer be able to activate key innate tumor suppressive mechanisms and instead of dying, as they would normally do, they are now able to survive and give rise to cancer. We highlight the importance of BCL-2 family members in tumor progression later in the chapter.
Oncogene Collaboration – from Cell Culture to Animal Models
It is the long history of humankind … those who learned to collaborate and improvise most effectively have prevailed.
Charles Darwin
An activating mutation in a single oncogene is not sufficient to cause cancer – rather oncogenes must collaborate with one another and with inactivation of tumor suppressors in order to generate cancers. Oncogene collaboration can be demonstrated in vitro using cultured cells in which various oncogenes are introduced, and also in vivo using genetically altered mice. In different cell types, different combinations of oncogenes are required for such collaborative events to transform normal cells into cancerous ones (transformation).
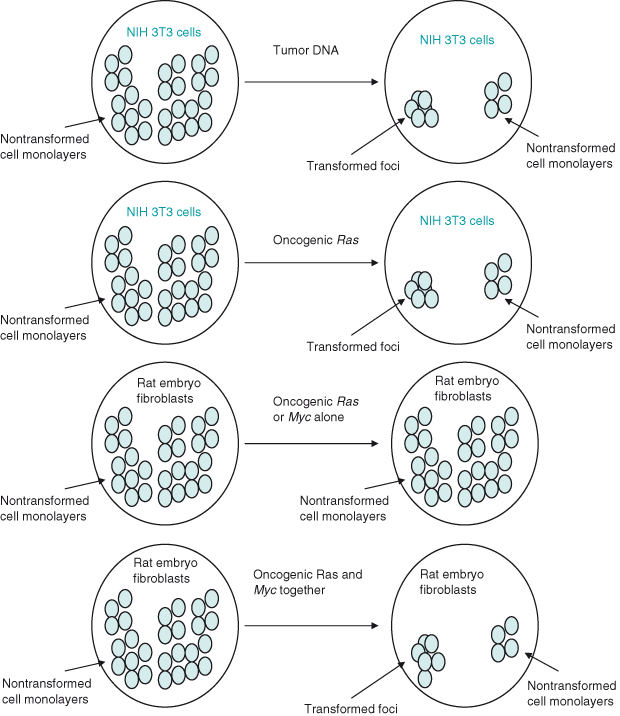
If normal (wildtype) rat embryo fibroblasts cultured in serum are transfected with the c-MYC oncogene alone or RAS oncogene alone, then they behave normally. However, when both oncogenes are transfected, the cells become transformed, that is, show various features or behavior associated with cancer cells (Fig. 6.3). These include enhanced proliferation and motility, and the ability to divide without adherence to a substratum, resulting in cells piling up on top of each other to form colonies. Although much important data has been generated, it is worth emphasizing again that the behavior of cells studied in vitro – in this case, to identify oncogenes that cause transformation – might not always be the same as when cells are studied within their normal context in an intact organism.
Figure 6.3 Oncogene collaboration in vitro. Transfection of NIH 3T3 fibroblasts with DNA from tumors or tumor cell lines results in some cells becoming transferred, as demonstrated by loss of normal contact inhibition and formation of foci (colonies) of cells piled up rather than in a monolayer. This same result could subsequently be achieved by transfecting NIH 3T3 cells with oncogenic RAS. In contrast, transfection of a “more normal” rat embryo fibroblast with any single oncogene (either RAS or MYC alone) did not result in transfomation. However, if both oncogenic RAS and MYC were transfected into rat embryo fibroblasts then foci did form – oncogene cooperation. By implication, NIH 3T3 fibroblasts have already undergone mutations such as those likely to occur in multistep tumorigenesis, thus enabling them to be more readily transformed in cell culture.
For this reason, genetically altered mice are studied in which expression of particular oncogenes is targeted to certain tissues and can be examined in the context of the whole animal. The oncogene is linked to a suitable promoter and then injected into a mouse egg nucleus. The oncogenic construct becomes integrated into the mouse genome leading to the generation of a strain of genetically altered mice that carry the oncogene in all cells (see Chapter 20). Depending on the promoter selected, expression of the oncogene will ultimately be driven in a specific cell type or tissue in the animal. Historical studies show that mice carrying either a c-MYC or a RAS oncogene linked to the MMTV promoter (which ultimately drives MYC or RAS overexpression in the mammary and salivary gland) develop tumors much more frequently than normal. However, when these two strains of mice are crossbred to generate mice carrying both c-MYC and RAS oncogenes, tumors develop at a far higher rate – the result of oncogene collaboration. Nevertheless, tumors arise only after a delay and in a small proportion of cells within the tissue. This implies that a further propitious genetic alteration is needed for tumors to develop in this particular tissue. Such an event is thus “acquired” in one or a few cells through chance. We will see later in this chapter, however, that there are examples of cell types within the mouse that require only two oncogenes for tumors to arise.
Further in vitro work in the early 1990s showed that if rat fibroblasts transfected with c-MYC oncogene alone are cultured in very low amounts of serum, instead of cells ceasing growth/replication (as they would normally do), they were driven to proliferate uncontrollably. Paradoxically, cells also started to die, with the end result being overwhelming cell death by apoptosis (Fig. 6.4). However, if the oncogene BCL2 was also overexpressed, then cells were rescued from death and allowed to proliferate excessively: BCL2 acts as an oncogene because the encoded BCL-2 protein inhibits apoptosis. These important findings gave birth to the concept that oncogenic mutations that drive uncontrolled proliferation (as is the case with c-MYC) also possess intrinsic tumor suppressing activity. In other words, cells that acquire such oncogenic mutations have a built-in “fail-safe” mechanism, ensuring that potential cancer cells will die by apoptosis rather than survive to accumulate further genetic mutations that may otherwise lead to cancer. Such a cell population would be unable to outgrow its environment unless apoptosis was inhibited.
Figure 6.4 c-MYC promotes cell death by apoptosis (in vitro). In the early 1990s, several laboratories made an intriguing discovery: oncoproteins such as MYC and the adenovirus E1A – both potent inducers of cell proliferation – were shown to possess apoptotic activity. Ectopic expression of MYC in fibroblasts that were cultured in the absence (or limited supply) of survival factors (e.g. factors that are present in fetal calf serum, such as the extracellular molecule, insulin growth factor-1 (IGF-1), that mediates cell survival via its receptor) led to apoptosis, with the eventual loss of the entire cell population. Although interpreted by some as a conflict of growth signals – oncogenes activate apoptosis if the proliferative pathway is blocked in some way – the most widely held view of oncoprotein-induced apoptosis is that the induction of cell cycle entry sensitizes the cell to apoptosis. In other words, cell proliferative and apoptotic pathways are coupled. However, the apoptotic pathway is suppressed as long as appropriate survival factors deliver antiapoptotic signals. In this scenario, the predominant outcome of these contradictory processes will depend on the availability of survival factors.
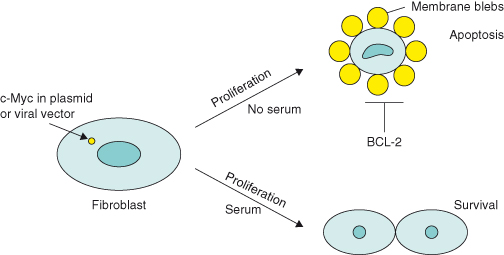
There is now compelling evidence in vivo to support this concept, as shown by the dramatic synergy between oncoproteins such as c-MYC and mechanisms that suppress apoptosis. An early example is the acceleration of lymphoma development in genetically altered mice that overexpress both c-MYC and the antiapoptotic protein BCL-2 in B lymphocytes, compared with mice expressing c-MYC alone. More recent mouse models include the overexpression of c-MYC together with an antiapoptotic protein, such as BCL-2 or BCL-XL, or loss of p19ARF or p53 tumor suppressors, some of which will be highlighted in this chapter. Later we will discuss another critical inherent tumor suppressing activity associated with oncogenic RAS, namely irreversible growth arrest (senescence).
A last important point to make is that such mouse models invariably have high levels of the particular oncogene(s) expressed. Recently, mouse models of cancer have been generated in which physiological (low) levels of oncogene expression more closely resemble the human disease process. These will be discussed later on.
The C-MYC Oncogene
Choices are the hinges of destiny.
Pythagoras
The c-MYC proto-oncogene family ranks among the most exhaustively studied groups of genes in biology not least because it is so profoundly involved in many human and other animal cancers.
The normal cellular proto-oncogene, c-MYC (MYC), encodes the protein c-MYC (MYC) whose key biological function is to promote growth and cell-cycle entry. However, as you will see later, this enigmatic protein appears to be a key player in a diverse, and sometimes opposing, group of cellular processes, including proliferation, growth, cell death (apoptosis), energy metabolism, differentiation, and angiogenesis (growth of new blood vasculature). A more recent function to add to the list is the central role played by MYC in the generation and maintenance of stem cells (see Chapter 9). From this point on we will refer to c-MYC/c-MYC (and all species versions of these) as MYC and MYC, removing the prefix c-, while for the other MYC family members the prefix (e.g. n-) will be retained.
Activated MYC Expression in Human Tumors
Our wretched species is so made that those who walk on the well-trodden path always throw stones at those who are showing a new road.
Voltaire
Before describing the mechanisms by which MYC activates or represses various biological processes in normal and in cancer cells, this section highlights what we know about MYC in human cancers and why we have had to look towards mouse tumor models in order to gain further insight into MYC’s oncogenic role in vivo.
MYC is expressed ubiquitously in cells during development and in growing tissues. Other homologs of MYC (L-MYC, N-MYC) are known (Box 6.2) and the expression of each is normally restricted to certain tissues. Likewise, in the adult, MYC is expressed in tissue compartments possessing high proliferative capacity (e.g. skin epidermis and gut), whereas it is undetectable in cells that have exited the cell cycle (quiescent). In human cancers, oncogenic MYC has long been known to be instrumental in the progression of Burkitt lymphoma. In this case MYC becomes oncogenic due to a translocational event (see Box 3.3), but in most cases where MYC contributes to human cancers this is actually not due to mutations in MYC but rather due to elevated or deregulated expression.
Activation of MYC has been detected in a wide range of human cancers, and is often associated with aggressive, poorly differentiated tumors. Such cancers include breast, colon, cervical, and small cell lung carcinomas, osteosarcomas, glioblastomas, melanoma, and myeloid leukemia. In most cases, the causes of MYC overexpression remain to be specifically described, but include gene amplification (see Table 6.2), whereby multiple copies of the MYC gene arise under the influence of genome instability (see Chapter 10), stabilization of MYC mRNA transcripts, enhanced initiation of translation due to mutation of the internal ribosomal entry site, or indirectly via activation of various upstream signaling pathways, such as WNT–β-catenin/LEF.
Levels of MYC protein are also influenced by control of degradation. Normal proliferating cells have evolved a number of mechanisms to limit the activity and accumulation of MYC. One of the most striking of these mechanisms is through the ubiquitin–proteasome pathway (see Chapter 11), which typically destroys MYC within around 20 minutes of its synthesis. Proteasome-mediated degradation of MYC is directed by at least two ubiquitin ligases – Fbw7 and Skp2. Interestingly, mutations in Fbw7 are noted in several types of tumors that manifest increased abundance of MYC. The ubiquitin-specific protease USP28 stabilizes MYC by interacting with Fbw7. USP28 is detected at high levels in colon and breast carcinomas and contributes to proliferation of these tumors.
Activation of MYC, as is the case in many human tumor cells, is defined as deregulation of the normal, highly controlled expression pattern of the MYC proto-oncogene. Until recently, however, distinguishing between cells that harbor normal or activated MYC has proved difficult. In normal dividing cells, MYC expression is maintained at a relatively constant intermediate level throughout the cell cycle, whereas in its oncogenic form, MYC might be constitutively expressed at levels ranging from moderate to very high, and is nonresponsive to external signals. Alternatively, the regulated pattern of MYC expression can remain intact, but exceed normal levels of expression for the given cell type.
Historically, molecular pathologists have relied on the presence of gross chromosomal abnormalities, such as translocation or amplification, of the MYC locus to define activation of this oncogene in tumor cells (as is the case in Burkitt lymphoma and neuroblastoma, respectively). However, this restrictive diagnostic criterion has resulted in an underestimate of the numbers of tumors with deregulated MYC. Therefore, the detection of MYC activation in tumor cells now relies on both elevated expression of MYC mRNA and genetic alterations of the MYC locus. Advances in technology allow us to evaluate expression levels of MYC mRNA in tumor cells (precisely excised using laser-capture microdissection) with real-time reverse transcription-polymerase chain reaction (RT-PCR) and expression profiling using microarray technology (see Chapter 18). These approaches allow rapid, quantifiable, high-throughput screening of MYC activation in tumor cells. However, these techniques do not measure levels of MYC protein expression, which are largely assayed by immunohistochemical methods.
The fact that MYC activation is present in a broad range of human cancers, and is often associated with a poor prognosis, suggests that analysis of MYC deregulation in certain tumor types may be used as a diagnostic marker. Moreover, inactivating MYC, or downstream targets of MYC, may represent useful therapeutic targets. To this end, much important information has derived from regulatable mouse tumor models (see later). These models have exposed various oncogenic properties of MYC during tumor progression as well as determining whether inactivating MYC in tumors can lead to their regression. Although still in its infancy, various approaches to target MYC in human tumors are presently under investigation. If MYC inhibition were even possible, what would be the consequences for the homeostasis of normal proliferating tissues versus the fate of cancer cells? This is a crucial question as, not surprisingly, a major challenge is achieving specific delivery of the MYC inhibitor only to the nucleus of tumor cells in vivo. Recent studies have shed light on this important issue and there is reason to be optimistic, as will be discussed later on in this chapter.
MYC – Transcription Factor and “Master Regulator” of the Genome
To rule is easy, to govern difficult.
Goethe
MYC is a transcription factor that requires dimerization with another protein, MAX, in order to become transcriptionally active (Fig. 6.5). Both MYC and MAX are basic helix–loop–helix zipper proteins (bHLHZ) and the MYC–MAX heterodimer binds the E-box sequence CACGTG of target genes to activate gene transcription – following the recruitment of multiple coactivator complexes, such as TRRAP (described in Fig. 6.5).
Figure 6.5 Functional domains of human MYC protein. (a) The C-terminal domain (CTD) of human MYC protein harbors the basic helix-loop-helix leucine zipper (bHLH-LZ) motif for dimerization with its partner, MAX, and subsequent DNA binding of MYC–MAX heterodimers. The N-terminal domain (NTD) harbors conserved “c-MYC boxes” I and II (MBI and MBII) essential for transactivation of c-MYC target genes. Recently, the MBIII situated in the central region has been found to be important for negatively regulating the apoptotic response. (b) Some major MYC-interacting proteins that may or may not bind simultaneously to MYC. These include coactivator TRRAP, part of a complex possessing histone acetylase activity (HAT), which interacts with the MBII region and mediates chromatin remodeling. TIP48 and TIP49 proteins interact with the NTD of MYC and are implicated in chromatin remodeling due to their ATP-hydrolyzing and helicase activities. Proteins involved in transcriptional regulation, such as MIZ-1, interact with the CTD of MYC, whereas SP1 interacts with the central region of MYC. NLS, nuclear localization signal.
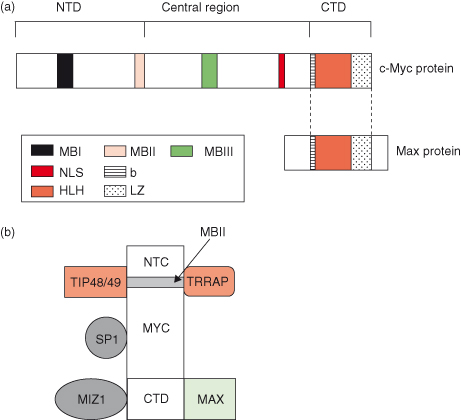
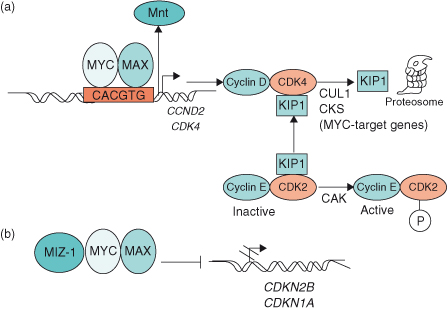
Figure 6.6 MYC induces cell cycle entry through activation and repression of target genes. (a) MYC–MAX heterodimers activate the target genes CCND2 (cyclin D2) and cyclin-dependent kinase 4 (CDK4), which leads to sequestration of CDK inhibitor KIP1 (p27) in cyclin D2–CDK4 complexes. Subsequent degradation of KIP1 involves two further MYC target genes, Cul-1 and Cks. In so doing, KIP1 is not available to bind to and inhibit cyclin E–CDK2 complexes, thereby allowing cyclin E–CDK2 to be phosphorylated by cyclin-activating kinase (CAK). Activation of some genes by MYC–MAX involves displacement of the putative tumor suppressor MNT from target genes. (b) MYC–MAX heterodimers repress the CDK inhibitors p15INK4B and WAF1 (p21), which are involved in cell cycle arrest. By interacting with the transcription factors MIZ-1 (and/or SP1), MYC–MAX prevents transactivation of INK4B (CDKN2B) and WAF1 (CDKN1A).
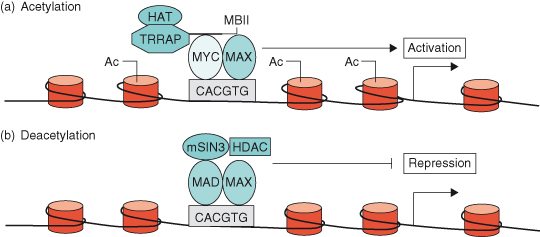
As discussed in Chapter 11 (see Box 11.1), DNA is not found in isolation in the cell but is associated with proteins called histones to form a complex substance known as chromatin. Thus, if chromatin is condensed, transcription factor complexes such as MYC–MAX cannot get to the DNA, and the genes will be switched off. Chromatin must be in an “open” conformation in order for gene transcription to occur and it is now known that the MYC–MAX transcription factor complex can lead to such a permissive alteration in the structure of chromatin – known as chromatin remodeling. For instance, TRRAP (transformation/transcription domain-associated protein), a MYC coactivator, is part of a complex that contains histone acetyl transferase (HAT) activity. Once recruited to MYC, TRRAP acetylates nucleosomal histone H4 at promoter sites, which alters the chromatin structure allowing accessibility of MYC–MAX complexes (see Fig. 6.7). Conversely, deacetylation of nucleosomal histones leads to switching off transcription of MYC target genes – a situation that often occurs during cellular differentiation when MAD levels are high (see Fig. 6.7b) (discussed later). Such chromatin remodeling is described as “epigenetic,” since modifications to the DNA or the histones have a huge impact on gene transcription and repression without changing the nucleotide sequence of the DNA. This important and fascinating topic is described in greater depth in Chapter 11.
Figure 6.7 MYC–MAX and MAD–MAX heterodimers regulate gene activation through chromatin remodeling. (a) MYC–MAX heterodimers binds to an E-box sequence (CACGTG) near the promoter of a c-MYC target gene. The coactivator TRRAP (transformation/transcription domain-associated protein), a component of a complex that contains histone acetyltransferase (HAT) activity, is then recruited to the MYC box II (MBII) domain of MYC and acetylates (Ac) nucleosomal histone H4 at the E-box and adjacent regions. Nucleosomal acetylation alters chromatin structure, allowing accessibility of MYC–MAX transcriptional-activator complexes to target DNA, resulting in expression of the target gene. (b) Induction of MAD during terminal differentiation results in the MAD–MAX heterodimer binding to an E-box of a c-MYC target gene. Corepressor SIN3 and histone deacetylases (HDACs) are then recruited to MAD, resulting in local nucleosomal histone deacetylation and repression of target gene expression.
While the pre-eminent role of MYC is to activate gene expression, there are, in fact, many important genes that are repressed by MYC – those involved in cell-cycle arrest, cell adhesion, and cell–cell communication. This makes biological sense if MYC is to drive the cell cycle during normal cell turnover and development, and will self-evidently also increase the threat posed by oncogenic deregulation in tumorigenesis. The powerful combination of driving expression of genes that promote the cell cycle (e.g. cyclin-dependent kinases – CDKs) while repressing those that inhibit the cell cycle (e.g. CDK inhibitors – CDKIs), allows tumor cells with deregulated MYC to replicate uncontrollably; MYC can press the cell-cycle accelerator and release the brakes at the same time.
Over the last few years, technological advances, such as expression profiling (microarrays) and genomic binding studies (see Chapter 20), have led to the surprising discovery that MYC possesses a large number of gene targets (100–1000s) which, depending on the cell type, amounts to 15–20% of all genes. No wonder that MYC has been described as a “master regulator” of the genome!
Intriguingly, despite MYC binding to promoter sites of thousands of genes, only a minority are actually transcribed by MYC. In this regard, with the use of high-throughput chromatin immunoprecipitation (ChIP) to identify binding sites, a model is emerging in which multiple transcription factors are required to fire off transcription. Even with these powerful technologies, the potential complement of MYC target genes operating at any given time in any given system is bewildering, with genes involved in diverse cellular functions.
Equally important has been the recent discovery that the outcome of MYC activation (i.e. which MYC target genes are transcribed or repressed) is acutely dependent on level of expression or activity within the cell. So, for instance, at low levels, MYC–MAX binds promoter-proximal E-boxes within CpG-rich (dinucleotide sequence, cytosine-phosphate diester-guanine), H3-K4-methylated (lysine methylation of histone H3) euchromatic islands, to transcribe target genes. (The linguistic skills required to decipher this sentence can be acquired in Chapter 11, where the relevance of such DNA/chromatin modifications is detailed.) Actually, when translated, this means simply that MYC will bind to promoter regions with a more open configuration of chromatin that is transcriptionally active. However, when expressed at very high levels, MYC can bind to promoters lacking an E-box! Furthermore, a ChIP-seq analysis of relatively high abundance MYC binding showed that while 62% of the most reliable sites possess E-boxes, nearly 40% of binding loci did not.
At present we know little about these non-E-box targets, but there is circumstantial evidence that these include RNA polymerase III-transcribed genes, such as the amino acid-bearing transfer RNAs (tRNAs). MYC associates with the TFIIIB subunit of the RNA polymerase III complex and mediates recruitment of acetyltransferase GCN5 (general control of amino acid synthesis protein 5) and the TRAPP complex described earlier. Therefore, the stimulation of polymerase III transcription is an example of MYC functioning as a coactivator, independent of its cognate DNA-binding site.
As the reader will by now be expecting, the all-pervasive microRNAs (miRNAs) are important in regulating MYC’s biological functions also. The regulation of miRNAs by MYC impinges upon multiple phenotypes that contribute to tumorigenesis – cell-cycle progression, apoptosis, metabolism, angiogenesis, and metastasis (see Fig. 6.8), and is discussed later under its own heading.
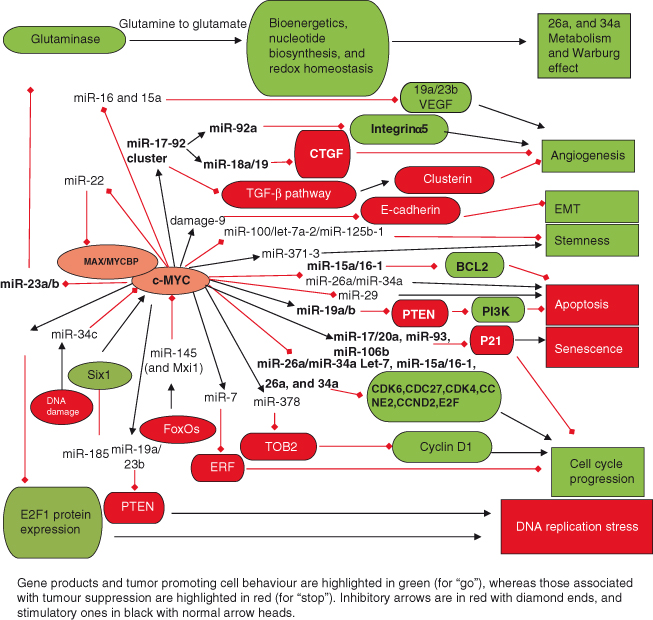
Figure 6.8 Role of miRNAs in regulating and mediating the actions of MYC. The first demonstration that miRNAs could have an important role in oncogenesis came with the discovery that MYC directly activates transcription of the miR-17-92 cluster to produce at least six mature miRNAs (miR-17, miR-18a, miR-19a, miR-19b-1, miR-20a, and miR-92-1), now known to be frequently amplified and/or overexpressed in many human cancers. As seen in the figure, MYC regulates an extensive network of miRNAs that affects many aspects of cell behavior, such as cell-cycle progression, apoptosis, metabolism, angiogenesis, and metastases. Only a few selected miRNAs are discussed here. Cell cycle: Both induction and repression of specific miRNAs by MYC facilitate transit through the cell cycle via complex mechanisms. For example, induction of miR-17 and related family members (miR-20a, miR-93, and miR-106b) promote cell-cycle progression by repressing levels of the cyclin-dependent kinase inhibitor P21 (CDKN1A), an essential protein involved in DNA damage/p53-mediated cell cycle arrest. Numerous miRNAs are repressed by MYC, including the let-7 family, known to exhibit antiproliferative, proapoptotic and/or anti-tumorigenic activities. For example, let-7 family members (miR-15a/16–1, miR-26a, and miR-34a) have a potent inhibitory effect on the cell cycle: miR-15a/16-1 inhibits expression of cell cycle regulators such as CDK6 and CDC27; miR-34a regulates CDK4, CDK6, CCNE2, and E2Fs, whereas miR-26a represses CCND2 and CCNE2. Thus, by downregulating expression of these miRNAs, MYC greatly facilitates cell-cycle progression. Cell survival/apoptosis: MYC’s upregulation of the miR-17-92 cluster inhibits apoptosis. In particular, miR-19 mediates cell survival by targeting the tumor suppressor PTEN, an antagonist of the PI3K–AKT survival pathway (see Chapter 7). Consequently, downregulation of PTEN leads to induction of PI3K signaling and prevention of apoptosis. In addition, transcripts of proapoptotic proteins such as BIM are targeted by miRNAs within the miR-17-92 cluster including miR-19a/b, thus inhibiting apoptosis. MYC also suppresses miRNAs to promote cell survival (e.g. the miR-15a/16-1 cluster that leads to downregulation of the antiapoptotic protein BCL-2). Angiogenesis: The miR-17-92 cluster plays a key role in angiogenesis. For example, miR-18a and miR-19 family members directly target negative regulators of angiogenesis such as CTGF (connective tissue growth factor) and TSP1 (thrombospondin-1), respectively. The reduced expression of these proteins ultimately allows angiogenesis to proceed. In contrast, the miR-92a member inhibits angiogenesis by targeting and thus downregulating the angiogenesis-promoting factor integrin α5. Future studies will shed light on whether specific members of the miR-17-92 cluster are present at different levels in specific cell types and/or cellular contexts and thus will dictate the phenotypic outcome. Metabolism: Glucose and glutamine metabolism are essential for cell survival and proliferation. MYC contributes to this activity through miRNA regulation: activation of the miR-17-92 cluster stimulates signaling through the PI3K–AKT pathway via downregulating expression of PTEN (described above). The PI3K–AKT pathway strongly promotes glucose metabolism and fatty acid synthesis, for example, by increasing glucose transporter surface expression and enhancing glycolytic enzyme activity. MYC promotes glutamine metabolism through several mechanisms, including direct transcriptional activation of glutamine transporters, as well as by repressing transcription of miR-23a/b, which target mitochondrial glutaminase (GLS). This leads to an increase in expression of GLS, which is a key enzyme that converts glutamine to glutamate – a substrate for the production of ATP.
To Cap It All, Now MYC Also Regulates Translation
The MYC proteins were initially identified as transcription factors that activate or repress specific protein-encoding genes transcribed by RNA polymerase II. As previously mentioned, we now know that they can activate the expression of tRNAs and rRNA via RNA polymerases I and III, as well as miRNAs (described later).
To this impressive portfolio we can further add a role in translation, as it has now been demonstrated that MYC is involved in mRNA cap methylation (see below). The biological ramifications of this newly discovered MYC function could be substantial, particularly, in light of the often modest transcriptional effects of MYC on target gene expression. Thus, only a 1.5- to 2-fold increase in mRNA is typically observed, whereas, by contrast, cap-methylated mRNA may increase by 3- to 6-fold. By this means, despite the modest transcriptional response, cap-methylated mRNAs can be translated into higher levels of protein.
RNA polymerase II transcripts require modification and processing before becoming mature transcripts that can subsequently be translated into proteins. For instance, exons are spliced together: the 3′ end of the transcript is cleaved, and polyadenylated RNA is exported from the nucleus to the cytoplasm, where translation factors bind to the RNA and direct recruitment of the ribosomal subunits needed for translation occurs. The methyl cap is an inverted 7-methylguanosine group joined to the first transcribed nucleotide by a triphosphate bridge, and is formed shortly after the initiation of transcription. The methyl cap – found at the 5′ end of mRNA – is crucial as it protects transcripts from premature degradation by exonucleases during translation. Thus it is essential for mRNA translation and cell viability.
So, at present we know that MYC promotes cap methylation of mRNAs corresponding to most of its target genes and, surprisingly, some that are not MYC target genes! Which other transcription factors possess such nontranscriptional activities? So far, E2F1 has been shown to enhance cap methylation on its target genes, but whether this ability extends to other transcription factors remains to be seen.
MYC – Cell Growth and Proliferation
No two men ever judged alike of the same thing, and it is impossible to find two opinions exactly similar, not only in different men but in the same men at different times.
Michel Montaigne
When a normal cell receives a signal to proliferate, such as that generated by binding of a growth factor ligand to its transmembrane receptor (see Chapter 5), signaling pathways inside the cell become activated and ultimately cascade that signal to the nucleus, leading to changes in gene expression. Among these changes will be the induction of specific genes required for cell-cycle entry, enabling the cell to leave its arrested state (G0) and re-enter the cell cycle (described in Chapter 4). The MYC proto-oncogene is one of the most important of these induced genes and is one of a select few so-called “early-response genes” (induced within 15 minutes of growth factor treatment in vitro). MYC plays a crucial role in allowing cells to exit G0 and progress through into S phase and proliferate. Conversely, when cells are not proliferating, MYC is silent. It is not difficult to picture the potential effect a gene like MYC might have on a cell, if it were continuously expressed or activated – that is, oncogenic MYC.
MYC is crucial for normal cell proliferation. Without it, cells would have a remarkably difficult time replicating themselves: normal development does not proceed in mice in which both Myc alleles have been inactivated, and embryos die in utero. In vitro, the cell-cycle effects of inactivating MYC have been investigated using a rat fibroblast cell line in which both alleles of c-MYC were ablated. Cells show greatly reduced rates of proliferation, mainly due to a major lengthening of G1 phase (4- to 5-fold) and the significantly delayed phosphorylation of the retinoblastoma protein (RB) (see Chapter 4). Thus, progression from mitosis at the end of one cell cycle to the G1 restriction point of the next cell cycle and the subsequent progression from the restriction point into S phase are both drastically delayed. Moreover, loss of MYC also leads to marked deficiency in cell growth (accumulation of mass), as demonstrated by a decreased global mRNA and protein synthesis.
Some studies in mouse knockdown models have suggested that at least under some circumstances cells may be able to replicate without normal levels of Myc, but this remains contentious as the extent of knockdown achieved and the proportion of cells affected is not clear and the role of other proteins, even other Myc family members, as substitutes for c-Myc under these circumstances is not known.
Normal healthy cells need to grow in size during the cell cycle before they divide. The first notion that MYC influenced cell growth came from the correlation between MYC and the expression of the rate-limiting translation initiation factors eIF4E and eIF2α, now known to be directly transcribed by MYC. Later studies showed that Myc has a direct role in the growth of invertebrate and mammalian cells: while diminished expression of the Drosophila ortholog of vertebrate myc (dmyc) resulted in smaller but developmentally normal flies, dmyc overexpression resulted in larger cells, with no significant change in division rate. A different outcome occurs in genetically altered mice when MYC is overexpressed in B lymphocytes: MYC induces cell growth but not cell-cycle progression in this cell type. Having said this, it is most likely that MYC induces both cell growth and proliferation in most cell types. It is indeed plausible that MYC’s role in regulating cell proliferation could at least in part be mediated through its effects on cell growth.
How does MYC mediate these effects on cell growth? Although the picture is not completely clear, RNA polymerase III (pol III) which is involved in the generation of tRNA and 5S ribosomal RNA (required for protein synthesis in growing cells), is activated by MYC via binding to TFIIIB, a pol III-specific general transcription factor (mentioned earlier). In fact, recent studies show that MYC can regulate the activity of all three known nuclear RNA polymerases and thus plays a central role in mediating ribosomal biogenesis and cell growth. In addition, as explained in the previous section, a new role of MYC recently discovered is its effect on translation via mRNA cap methylation – leading to an increase in the levels of protein synthesis.
Over the last decade it has become clear that MYC can drive several key metabolic pathways required to support cell growth: glycolysis, biosynthetic pathways, mitochondrial function, and glutaminolysis. Some of this has already beeen discussed in Chapter 5 under the heading of cancer cell metabolism and the Warburg effect.
Insights into how MYC might promote cell proliferation have resulted from a number of important studies revealing MYC’s ability to activate or repress target genes involved in cell-cycle progression (Fig. 6.6). As described in Chapter 4, progression of the cell cycle from G1 phase to S phase (DNA synthesis) is controlled by the activities of the cyclin-dependent kinase (CDK) complexes: cyclin D–CDK4 and cyclin E–CDK2. MYC induces cyclin E–CDK2 activity early in the G1 phase of the cell cycle, which is regarded as an essential event in MYC-induced G1/S progression. Importantly, both CCND2 (which encodes cyclin D2) and CDK4 are direct target genes of MYC, and their expression leads to sequestration of the cyclin-dependent kinase inhibitor (CKI) p27KIP1 by cyclin D2–CDK4 complexes. The subsequent degradation of p27KIP1 involves two other MYC target genes – CUL-1 and CKS. Therefore, by preventing the binding of p27KIP1 to cyclin E–CDK2 complexes, MYC allows inhibitor-free cyclin E–CDK2 complexes to become accessible to phosphorylation by cyclin-activating kinase (CAK) (see Fig. 6.6). As a consequence, increased CDK2 and CDK4 activities would result in RB hyperphosphorylation and subsequent release of E2F from RB (see Chapters 4 and 7).
Other important studies support the idea that MYC may also exert important influences on the cell cycle by repressing genes, such as those encoding the CKIs p15INK4b and p21CIP1, which are involved in cell-cycle arrest (see Chapters 4 and 7), through the MYC–MAX heterodimer interacting with transcription factors such as MIZ-1 and SP1 (Fig. 6.6). Consequently, the interaction of MYC–MAX with MIZ-1 blocks the association of MIZ-1 with its own coactivator (P300 protein), with the subsequent downregulation of p15INK4b, p21CIP1, and p57kip2. How important then, is MYC’s interaction with MIZ-1 in promoting tumor development? It has now been shown in transgenic mouse models that such interactions are critical for the induction and maintenance of T-cell lymphomas. Transgenic mice expressing a mutant form of MYC
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


