Chapter 2 Development of the lungs; perinatal and developmental lung disease
The traditional division of disease into congenital and acquired is less useful in the case of the lungs than one that considers developmental as opposed to non-developmental disorders. This is because lung development continues long after birth. Developmental disease may therefore be acquired well into the postnatal period. Conversely some congenital lung disease is non-developmental but acquired in utero, a good example of which is congenital pneumonia. Other abnormalities may be acquired in utero because of adjacent developmental anomalies, one example of which is a defect in the diaphragm admitting abdominal viscera into the thorax and thereby causing pulmonary hypoplasia.
Development of the lungs
Airway development
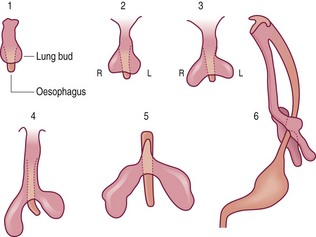
Human lung development is first marked by a longitudinal groove that appears on the ventral side of the primitive foregut 26 days after fertilisation. This separates from the foregut, first at its caudal end, and then progressively forward until a connection is retained only at the cranial end. This bud then bifurcates and grows on either side of the foregut as the embryonic lungs, with successive branching giving rise to the lobar (about 33 days), segmental and further divisions of the airways (Fig. 2.1).1 The developing lungs protrude into the coelomic cavity, the mesothelial lining of which forms from the mesoderm about the 14th day of gestation. With the development of the diaphragm and a pleuropericardial membrane the lungs become confined to the pleural cavities (Fig. 2.2).
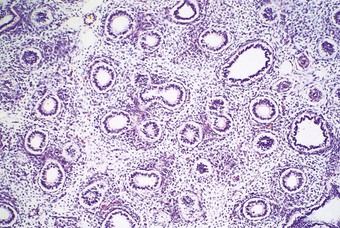
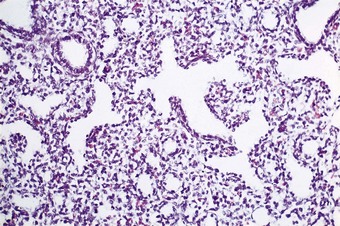
The phases of lung development and the major events therein are summarised in Table 2.1. The embryonic stage of lung development lasts until 6 weeks’ gestation, by which time the lungs have acquired a pseudoglandular appearance (Fig. 2.3). At this time the primitive potential air spaces are widely separated by abundant mesenchyme and lined by a vacuolated columnar or cuboidal epithelium rich in glycogen. The glycogen is particularly plentiful in the budding terminal epithelium where mitoses are most frequent. The pseudoglandular stage of development lasts until 16 weeks by which time division of the conductive airways down to the terminal bronchiole is complete.2 This is followed by a canalicular stage marked by the appearance of further air spaces which have an attenuated lining epithelium (Fig. 2.4). During this period the epithelium loses its glycogen and differentiates into the type I and II cells seen in adult alveoli. Although no proper alveoli are yet present, respiration is possible towards the end of the canalicular period around 28 weeks. Substantial amounts of connective tissue still separate the air spaces at this time but in the saccular stage of intrauterine lung development the connective tissue progressively diminishes by an apoptotic mechanism3 as new air spaces called terminal sacs develop. These resemble adult alveoli but are shallower and their walls still have enough connective tissue to separate the capillaries abutting air spaces into two layers. The development of true alveoli, first apparent at 36 weeks’ gestation, is characterised by infolding of one of the two capillary layers, further thinning of the connective tissue and the eventual fusion of the double capillary layer into one.4
Table 2.1 Major events during lung development
| Phase | Gestation | Major events |
|---|---|---|
| Embryonic | Up to 6 weeks | Lung buds form Segmental airways appear (6 weeks) |
| Pseudoglandular | 6–16 weeks | Bronchial smooth muscle appears (7 weeks) Neuroendocrine cells and submucosal glands appear (8 weeks) Bronchial cartilages appear (10 weeks) Airway branching complete (16 weeks) |
| Canalicular | 17–28 weeks | Basal cells appear (17 weeks) Clara cells appear (25 weeks) Basic structure of the acinus develops Mesenchyme starts to regress Blood vessels grow into acinar mesenchyme Type II alveolar cells appear (22 weeks) |
| Saccular | 29–35 weeks | Subdivision of saccules Further thinning of mesenchyme |
| Alveolara | 36 weeks to term | Terminal air spaces lined by flat epithelial cells Inconspicuous interstitium Double layer of alveolar capillaries fuses to give a single layer |
| Birth | Lung liquid rapidly cleared |
a Alveolar multiplication continues from 36 weeks well into childhood.
Alveoli develop mainly in the first year of extrauterine life and lung disease in infancy seriously impairs this. At birth, there are about 24 million terminal sacs and alveoli, compared to the adult figure of about 300 million alveoli. In the first year of life there is a fivefold increase in alveolar number with continued but slower increase thereafter, up to about 8 years of age and perhaps well into adolescence.5 There is also remodelling of airways in the first year of life. This entails an increase in length of all airway generations, further branching of alveolar ducts to give more generations, transformation of distal respiratory bronchioles into alveolar ducts by increasing alveolisation of their walls, and transformation of terminal into respiratory bronchiole by centripetal alveolisation.6–8 An important facet of postnatal lung development is that events in infancy and childhood may predispose to pulmonary disease in adult life. Thus, an increased frequency of respiratory infections in childhood has been identified in adults with chronic airflow obstruction.9–11
All the epithelial tissues of the lung, down to and including the alveoli, are of endodermal origin and all the interstitial elements are mesodermal in origin. This is contrary to older views on the development of the lung which envisaged the peripheral parts being purely mesodermal, but is demonstrated quite conclusively by electron microscopic studies of the developing mammalian lung. These show that the epithelial cells of the primitive endodermal bud extend continuously down to the smallest air spaces and that the epithelial lining of these spaces is not contributed to by any mesodermal elements.12–14
Vascular and neural development
The blood supply to the developing lung buds is derived from the sixth pair of aortic arches. On the right, the ventral part of this arch persists as the right pulmonary artery whilst the dorsal part disappears. On the left, the ventral part persists as the ductus arteriosus. Within the lung buds blood vessels are formed by two processes. Angiogenesis (the sprouting of new from pre-existing vessels) underlies the formation of proximal lung vessels whilst vasculogenesis within the mesenchyme (the formation of vascular lakes by precursor angioblasts) contributes the peripheral vascular component, the two ultimately fusing by a lytic process.15,16 The pulmonary arteries develop alongside newly formed airways, the latter seemingly acting as templates for the arteries.17 A well-defined branching pattern is apparent by 14 weeks’ gestation and all preacinar arteries are present by 16 weeks. At 20 weeks the branching pattern resembles that of adults. However, many peripheral arteries develop after birth (Fig. 2.5).18
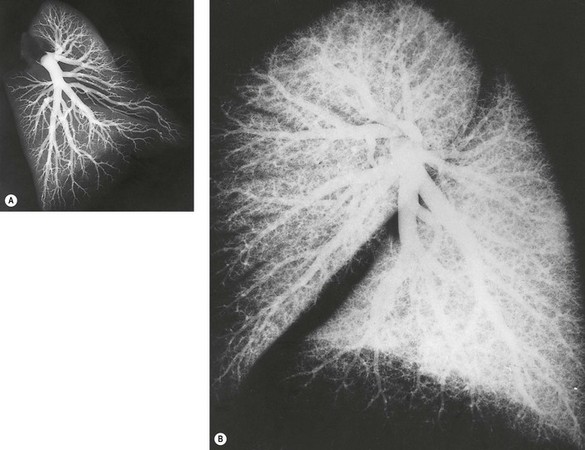
The processes of vasculogenesis and angiogenesis are also involved in the development of the pulmonary veins.19 Veins and arteries both first develop close to the primitive lung bud but the veins gradually take up a more peripheral position and by the time that the lung has a lobular structure the primitive veins are found in their adult position between the lobules. They initially enter a plexus around the foregut but later switch to a primary pulmonary vein that grows from the developing left atrium towards the lung buds. Incorporation of the primary pulmonary vein and its first two orders of branches into the wall of the left atrium results in two pulmonary veins from each lung entering the left atrium separately. Failure of the intrapulmonary veins to switch from the enteric venous plexus to the primary pulmonary vein underlies the congenital condition of anomalous pulmonary venous drainage (see p. 76).
Neural tissue derived from the vagus nerve extends into the growing tips of the fetal airways and bronchial smooth muscle, which develops from 7 weeks’ gestation, contracts in response to nervous stimulation from early gestation.20
Factors controlling lung development
Molecular mechanisms play an important role throughout lung development from the early induction of tracheal separation from the embryonic oesophagus to the appearance of cells specific to the lung such as the bronchiolar Clara cell and the alveolar pneumocytes. Various members of the forkhead homologue hepatocyte nuclear factor-3 (HNF-3) family are essential for the formation of the foregut endoderm and derivative organs, including the lung, while the induction of tracheal budding is dependent upon the simultaneous expression of the transcription marker Nkx2.1 (also known as thyroid transcription factor-1 or TTF-1).21–23 TTF-1 is expressed throughout the embryonic lung in the early stages of its development but later becomes restricted to the more distal elements where growth is most active. It is particularly involved in epithelial cell differentiation.
The endodermal and mesodermal elements of the lung are each important to the proper development of the other with the basement membrane, cell adhesion factors such as fibronectin and integrin, and a variety of growth factors all playing a role in this interaction.24–30 Growth factors include epithelial signalling molecules such as bone morphogenetic factor, transforming growth factor and sonic hedgehog that influence the underlying mesenchyme while the mesenchyme expresses signalling molecules such as fibroblast growth factor that are important in pulmonary epithelial development. Contacts between interstitial and type II epithelial cells have been identified in the newborn and shown to diminish during a postnatal proliferative phase.31 Experiments demonstrate that respiratory epithelium has an inhibitory effect on the growth of the underlying mesenchyme,32,33 whilst in fetal organ culture, intestinal mesoderm will induce bronchial budding but not branching, bronchial mesoderm will induce the tracheal endoderm to branch, and tracheal mesoderm will inhibit bronchial branching.34 Furthermore, whilst the bronchial mesoderm of one species will induce the endoderm of another to branch, the pattern of endodermal branching is that of the species contributing the mesoderm. These experiments clearly demonstrate the interdependence of mesoderm and endoderm in pulmonary development. They are reinforced by the arrested development of endodermal lung buds within ectopic neuroglia (see p. 73).
The fetal air spaces are filled by a chloride-rich fluid secreted by their lining epithelium, which makes a substantial contribution to the production of amniotic fluid.35 Its presence in the airways appears to be important to fetal lung growth for this is retarded if the fluid is drained.36 Conversely, ligation of the trachea results in large lungs.37
Neuroendocrine cells are particularly numerous in the respiratory epithelium of the developing airways38–42 and at least one of their products, gastrin-releasing peptide (human bombesin), is known to stimulate lung growth and maturation during the later stages of development.43 The expression of gastrin-releasing peptide receptor gene peaks at a time and at sites of most rapid airway growth and development.44
Respiratory movements take place in utero and these too are thought to be important for fetal lung growth and maturation.45 Section of the cervical cord above, but not below, the phrenic nucleus leads to severe pulmonary hypoplasia.46
Hormones are also important in lung development. The lungs bear steroid receptors, and maturation, but not growth, is promoted by glucocorticosteroids and probably other hormones. Glucocorticoid treatment of the fetus accelerates epithelial glycogen depletion, differentiation into type I and II cells, the appearance of lamellar bodies in type II cells, and their release into the air spaces.47 This is utilised in clinical practice to minimise the risk of respiratory distress in premature infants (see below): a decreased incidence of the respiratory distress syndrome and increased survival are reported in premature infants exposed to maternal steroid therapy.48 The steroids appear to act, at least in part, by stimulating lung fibroblasts to produce an oligopeptide known as fibroblast pneumonocyte factor which in turn stimulates surfactant synthesis by the type II cells, an example of paracrine activity.49
Perinatal disorders
Meconium aspiration
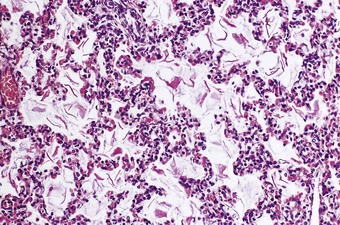
The expulsion of meconium into the amniotic fluid occurs as a response to fetal stress from a wide range of causes and its aspiration may cause a chemical or infective fetal pneumonia,50 described below under neonatal pneumonia. Fetal stress also increases the normal respiratory excursions that take place before birth and may result in the airways being obstructed by numerous fetal squames and mucus (Fig. 2.6). Pathologists are likely to encounter only fatal cases but the lungs are inherently normal and the obstruction is potentially reversible. The natural clearance mechanisms can be augmented with suction but such treatment may need to be prolonged, in which case oxygenation of the blood has to be maintained with an extracorporeal device; mechanical ventilation would only impair clearance. The mortality remains high, although corticosteroid and surfactant treatment are of some benefit.51–53
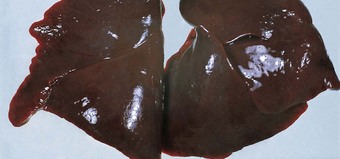
Massive pulmonary haemorrhage
This is a fairly common finding in perinatal necropsies and has several causes. In some cases of apparent pulmonary haemorrhage the findings are due to heavy blood-staining of oedema fluid rather than true haemorrhage and in these patients shock and left ventricular failure are important contributory factors. A coagulopathy, usually acquired, may cause true haemorrhage but the commonest cause is asphyxia. Cerebral oedema and haemorrhage are often also present and probably represent further manifestations of asphyxial damage. The condition is disproportionately frequent among preterm babies with very low birth weights54 and it is postulated that abnormal alveolar surface forces may contribute to the bleeding.55
Neonatal pneumonia
With transplacental transmission the pneumonia is only part of a generalised infection. Causative agents include cytomegalovirus, rubella virus, herpes simplex virus, varicella virus, mycoplasmas, Listeria monocytogenes, Treponema pallidum, Mycobacterium tuberculosis and Toxoplasma gondii. Many of these microbes elicit specific reactions that are dealt with in later chapters. However, it may be mentioned here that eosinophilic nuclear inclusions that represent parvovirus may be found in the lungs and other organs of premature stillbirths and neonates with non-immune hydrops.56
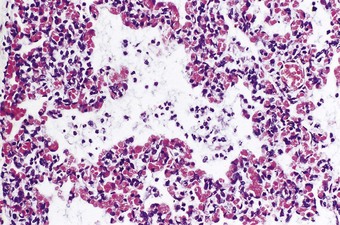
Pneumonia acquired by aspiration of infected amniotic fluid is known as congenital or intrauterine pneumonia. It is particularly likely if there is prolonged rupture of the membranes. Chorioamnionitis suggests this mechanism and examination of the placenta and its membranes is therefore helpful. Vaginal group B haemolytic streptococci or Candida albicans may be responsible, as may bowel bacteria such as Escherichia coli, klebsiellae and Pseudomonas aeruginosa. The air spaces contain polymorphonuclear leukocytes and fibrin but neither of these is as prominent as in an adult with pneumonia (Fig. 2.7). The distribution of the process is that of a bronchopneumonia. In some cases there is a combination of hyaline membrane disease and pneumonia. Bacteria may then be seen in the hyaline membranes, rendering them haematoxyphilic. It is possible that in such cases the bacteria promote the formation of the membranes by damaging the alveolar epithelium. Acute pneumonia and hyaline membranes may also result from the chemical toxicity of the bilirubin in aspirated meconium.50 The fact that atelectasis is minimal helps to distinguish pneumonia with hyaline membranes from the infantile respiratory distress syndrome, which is dealt with next.57 The combination of pneumonia and hyaline membranes is also seen in immature babies requiring ventilator support when there is secondary infection.58,59
Infantile respiratory distress syndrome (hyaline membrane disease)
By 28 weeks’ gestation, the alveolar epithelium has differentiated into type I and type II pneumocytes, surfactant can be detected and an extrauterine existence is possible. With modern intensive care, an increasing number of babies born before 28 weeks are surviving, but without it the chances of successful respiration at this age are slim. By 34–36 weeks, premature birth is generally followed by successful respiration but the capacity of type II pneumocytes to replenish spent stocks of surfactant is frequently inadequate, in which case respiratory distress becomes evident within a few hours of birth.60 This is apparent as grunting, indrawing of the intercostal tissues on inspiration and hypoxia. Radiographically there is opacification of the whole lung fields except for an ‘air bronchogram’. Without respiratory support the death rate in the first few days is high.
Pathological findings
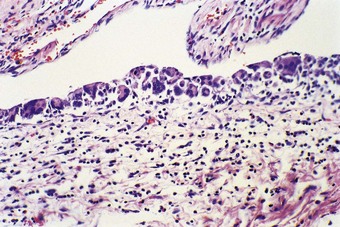
At necropsy the lungs are heavy, dark and airless (Fig. 2.8). Microscopy confirms the atelectasis, with air limited to the bronchioles (Fig. 2.9). There is also lymphatic distension and interstitial oedema, best seen in the perivascular connective tissue sheaths (see Fig. 2.9). Alveolar collapse and interstitial oedema are the expected consequences of high surface tensive forces acting on the alveolar walls. Hyaline membranes are found at the boundary of the air-filled bronchioles and the collapsed alveoli (Fig. 2.10). They are not found when death occurs in the first few hours of life and they are no longer thought to play a causal role in the alveolar collapse. The hyaline membranes represent compacted plasma exudates and cellular debris. They do not stain for fibrin with conventional histological stains but fibrin can be demonstrated in them by immunocytochemistry and epithelial necrosis by electron microscopy.61 The hyaline membranes are yellow if there has also been unconjugated hyperbilirubinaemia (Fig. 2.11).62,63
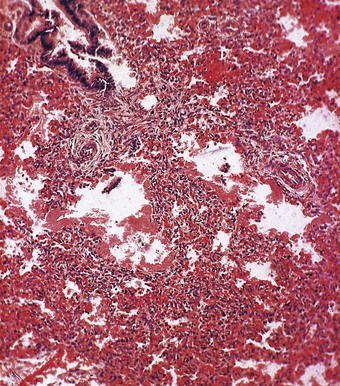
Hyaline membranes are not specific to premature babies and may be found in term infants who die of birth asphyxia some hours later. Virtually identical pathological changes are seen in the adult respiratory distress syndrome (see p. 135). Hyaline membrane disease is evidently a consequence of non-specific injury to the bronchiolar and alveolar lining. In premature infants the injury is likely to be physical; ventilatory efforts in the surfactant-deficient lung put undue shear forces on the epithelium at the air–liquid interface, which at end-expiration is at the bronchiolar level, the alveoli having collapsed completely. Such mechanical stress operates during each ventilatory cycle so that it is repeated several thousand times in the first few hours of extrauterine life.61
Therapy and its complications
If premature birth is expected, or induction of premature labour planned, the risks of respiratory distress can be reduced by administering corticosteroids to the mother.48 This promotes maturation of the fetal lung, including surfactant secretion.47 It is a preventive measure that is only feasible when premature birth can be anticipated.
After birth, mechanical respiratory support with oxygen enrichment is the main means of maintaining oxygenation. This is being increasingly supplemented by the insufflation of natural or synthetic surfactant into the infant’s airways.64 Such surfactant therapy is proving very successful but, if the infant does come to autopsy, the exogenous surfactant is seen in the air spaces as a yellow birefringent material. Another useful measure is the administration of nitric oxide gas in low concentrations (less than 80 parts per million) to induce pulmonary arterial dilatation and improve ventilation/perfusion matching. Extracorporeal oxygenation of the infant’s blood may also be undertaken but results are best in babies greater than 37 weeks’ gestation: preterm babies weighing less than 2 kg have blood vessels that are too fragile to withstand the damage from cannulae and are at risk of ventricular haemorrhage from the heparinised circuits.
With these measures the survival rate of preterm infants who develop respiratory distress has been greatly increased and hyaline membrane disease is now seen far less frequently. However, there are cardiopulmonary complications of such treatment. Immediate complications of positive-pressure ventilation include interstitial emphysema and pneumothorax (see below), while patency of the ductus arteriosus and cardiac failure (persistent fetal circulation, see below) may dominate the clinical picture after a few days. Some infants who survive the first weeks of life on ventilator support develop diffuse lung disease and die in the succeeding months from extensive bronchopulmonary dysplasia (see below). Long-term survivors tend to have impaired lung function for some months65,66 and a high incidence of bronchiolitis and pneumonia in infancy.67 Babies who recover spontaneously from the respiratory distress syndrome do so completely and suffer none of these pulmonary complications.
Bronchopulmonary dysplasia and chronic lung disease of prematurity
Bronchopulmonary dysplasia is the condition found in premature infants who are enabled with the help of artificial ventilation to survive the respiratory distress syndrome but die in the first few months of life.68,69 As infants who recover spontaneously do not develop this condition it must be attributed to the respirator support but it is controversial whether oxygen toxicity or barotrauma is mainly responsible.70,71 Other factors that have been incriminated include prematurity, infection, tracheal intubation, patency of the ductus arteriosus and fluid overload.
It represents disordered prolongation of the healing phase of hyaline membrane disease in which the original damage is augmented by the supportive therapy. Interstitial oedema persists and granulation tissue develops in the walls of the airways and alveoli, progressing to interstitial fibrosis. The air–blood barrier is thickened and the alveolar capillaries are reduced in number. There is also organisation of the hyaline membranes, and intraluminal fibrosis leads to the obliteration of many bronchioles. Regenerative hyperplasia is evident in the epithelium of alveoli, bronchioles and bronchi, often with metaplastic changes (Fig. 2.12). Distal air spaces are lined by cuboidal type II pneumocytes or atypical elongated cells which represent type II cells differentiating into type I. Severe squamous metaplasia is often evident in the larger airways. The epithelial changes resolve with time and in babies who survive longer than 3 months the changes are more marked in the lung parenchyma. The changes are often patchy and consist of fibrotic airless areas of lung tissue alternating with distended or even emphysematous areas (Fig. 2.13). Fibrosis predominates in babies who die in the first 2 months and emphysema in those who survive a year or more.72 Long-term survivors generally have impaired lung function.73 They also show hypertrophy of the right ventricle.74
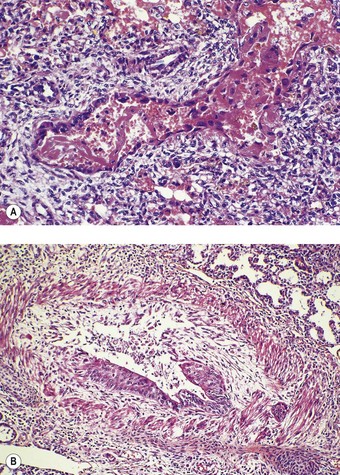
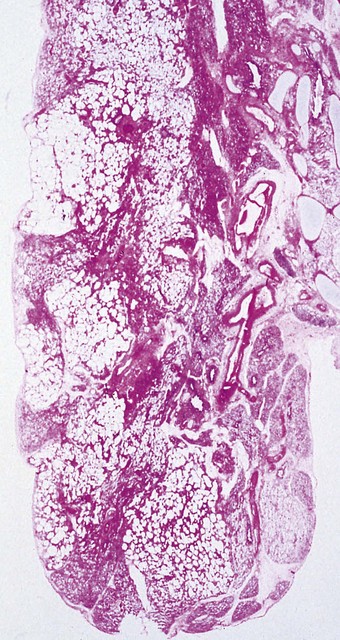
Figure 2.13 Bronchopulmonary dysplasia. Solid areas of collapse and fibrosis alternate with lighter distended areas.
(Courtesy of Dr W Geddie, formerly of Toronto, Canada.)
Chronic lung disease of prematurity is characterised by large simple air spaces lined by undifferentiated cuboidal cells and separated by cellular fibroelastic septa of even thickness. Mean linear intercept counts are low, indicating reduced numbers of air spaces and arrested alveolar development. The squamous metaplasia and alternating areas of collapse and overinflation seen in bronchopulmonary dysplasia are not well represented in chronic lung disease of prematurity (Box 2.1).75,76
Box 2.1 Modified from Coalson76 with permission of Elsevier.
A comparison of the pathological features of bronchopulmonary dysplasia and chronic lung disease of prematurity (‘Old and New BPD’)
Bronchopulmonary dysplasia (‘Old BPD’)
Alternating areas of hyperinflation and atelectasis
Severe airway epithelial lesions (hyperplasia and squamous metaplasia)
Decreased internal surface area and alveoli
Airway smooth muscle hyperplasia
Chronic lung disease of prematurity (‘New BPD’)
Deceased large, simplified alveoli (alveolar hypoplasia, decreased alveolar complexity)
Negligible airway epithelial lesions
Variable airway smooth muscle hyperplasia
Variable interstitial fibroproliferation
Decreased, dysmorphic capillaries
Less severe vascular hypertensive lesions
Surfactant dysfunction disorders
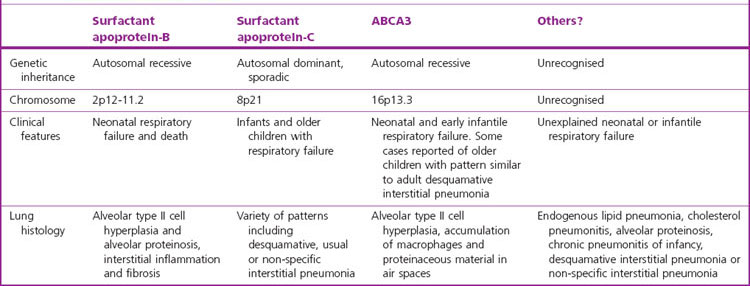
The surfactant deficiency underlying the great majority of cases of the infantile respiratory distress syndrome (hyaline membrane disease) is attributable to immaturity of the lung but the syndrome is also seen in a few mature babies who have an inherited defect in surfactant production.77 To date, three such conditions have been identified in humans, respectively involving surfactant proteins B and C and an integral membrane protein known as ABCA3 (Table 2.2).78
The best known is that involving faults in surfactant protein B synthesis.79–82 Various mutations of the surfactant protein B gene have been identified, resulting in considerable molecular and phenotypic variability.83–85 These babies may die with hyaline membrane disease or bronchopulmonary dysplasia or survive the neonatal period only to develop changes that resemble pulmonary alveolar proteinosis, desquamative interstitial pneumonia or cholesterol pneumonitis together with type II cell hyperplasia, features that overlap with those of chronic pneumonitis of infancy, described below.79,86 It seems likely that in protein B deficiency the alveolar proteinosis is due to excessive amounts of surfactant being secreted in order to compensate for its defective protein content. As in autoimmune alveolar proteinosis, there is an initial stage of endogenous lipid pneumonia, representing macrophage ingestion of the excess surfactant. Electron microscopy shows abnormal type II cell inclusions: these lack their normal whorled lamellae and resemble those described in mice in which the surfactant protein B gene has been disrupted.87,88 The diagnosis of surfactant protein B deficiency may be established by immunostaining bronchoalveolar lavage material or lung tissue for the various surfactant proteins and thereby demonstrating an absence of surfactant protein B and increased amounts of surfactant protein A.
Abnormalities involving surfactant protein C have also been reported,89 again with considerable phenotypic variation.90 Most patients have been neonates or infants but some have been older children or adults. Young children generally show the changes of chronic pneumonitis of infancy described below,91 but some show alveolar proteinosis whilst other cases have been adults displaying histological patterns of usual desquamative or non-specific interstitial pneumonia.90
An absence of surfactant lamellar bodies from type II alveolar cells in term infants who develop respiratory distress soon after birth represents a further genetic error in surfactant production, one involving an integral membrane protein known as ABCA3.92–95 This is an ATP-binding cassette protein, one of several concerned in transmembrane delivery of a variety of substances, including lipids. In the lung it has been localised to the limiting membrane of the surfactant lamellar bodies. ABCA3 deficiency is associated with a histological pattern of desquamative interstitial pneumonia, non-specific interstitial pneumonia, endogenous lipid pneumonia, alveolar proteinosis or, rarely, usual interstitial pneumonia.92,93,96,97 A variant of ABCA3 deficiency is characterised by the presence of abnormal lamellar bodies that have been likened to fried eggs.98 Genetic studies have subsequently identified ABCA3 deficiency in older children and young adults with a variety of obscure interstitial lung diseases.95,99
It is likely that children reported in the older literature as showing a combination of endogenous lipid pneumonia, cholesterol granulomas and alveolar proteinosis of obscure etiology represented further cases of abnormal surfactant production, possibly due to as-yet unrecognised defects.100–105 Deficiency of surfactant protein A has not yet been firmly established in humans but may be represented by the Hermansky–Pudlak syndrome described on page 491, and the pale ear mouse, which provides an animal model of the human syndrome, where it appears that faulty production of surfactant protein A leads to the development of giant lamellar bodies and pulmonary fibrosis.106,107
Chronic pneumonitis of infancy
Chronic pneumonitis of infancy represents a pattern of disease first described in full-term infants of either sex whose mothers experienced uncomplicated pregnancies and deliveries.108 The infants are normal at birth but cough, respiratory distress and a failure to thrive develop at ages varying from 2 weeks to 11 months (average 3.6 months). Radiographs show a predominantly interstitial pattern of opacification. The prognosis is variable: some of the children recover spontaneously whereas others die of respiratory failure within 3 months of the onset of symptoms.
The aetiology is unknown and may be multifactorial. It has been suggested that the condition represents recurrent or poorly cleared infection108 but there is no firm evidence of this. The changes within the air spaces often resemble those of alveolar proteinosis, suggesting that causes of this disease in infancy such as surfactant protein B or C deficiency, as described above, and lysinuric protein intolerance109–111 may be responsible. Gastro-oesphageal reflux with aspiration is another possibility.
Biopsy shows alveolar wall thickening with a variety of air space abnormalities. There is little inflammation. The alveolar wall thickening is due to interstitial spindle cell proliferation and type II epithelial cell hyperplasia. Collagen is not a feature in the early stages but the disease may progress to interstitial fibrosis. Within the alveolar lumen, macrophages are increased and there is often an accumulation of granular eosinophilic material in which acicular cholesterol crystal clefts may be seen. The macrophages may have foamy cytoplasm (Fig. 2.14).
Cellular interstitial pneumonitis and pulmonary interstitial glycogenosis
Cellular interstitial pneumonitis shows less marked interstitial thickening, type II cell hyperplasia and alveolar filling than chronic pneumonitis of infancy and appears to have a better prognosis. However, whether this means that the two represent separate conditions91 or are variations of one112,113 is debatable.
Some of those that distinguish cellular interstitial pneumonitis and chronic pneumonitis of infancy equate the former with what has been described as pulmonary interstitial glycogenosis,91 a term that was coined when vacuoles in the interstitial cells were shown to represent glycogen (Fig. 2.15).114 Such glycogenosis appears to be self-limiting115 and may be merely an epiphenomenon, as interstitial cells with clear cytoplasm are not infrequently seen in association with other disorders such as antibody deficiency116 and alveolar capillary dysplasia.117 The term ‘glycogenosis’ is unfortunate as it usually refers to an inborn error of glycogen metabolism due to enzyme deficiency for which there is no evidence in this condition: the term ‘pulmonary interstitial glycogen accumulation disorder’ is therefore preferred by some authors.118
Interstitial emphysema
This condition is a form of barotrauma and in infancy is often a complication of mechanical ventilation. High ventilatory pressures rupture the alveolar walls and permit air to enter the interstitial tissue. The air is seen particularly in the abundant connective tissue that surrounds the pulmonary artery and bronchiole in the centre of the acinus and forms the interlobular septa. Lines of bubbles are seen through the pleura, outlining the lung lobules. Blebs may project from the pleura, and cyst-like spaces up to 3 cm in diameter are found on the cut surface of the lung (Figs 2.16 and 2.17).119–121 Pneumothorax and surgical emphysema of the mediastinum and neck frequently accompany interstitial emphysema. The air may track into the other serosal cavities causing pneumopericardium or pneumoperitoneum. Rarely, large subpleural cysts develop.122 In some cases the changes resolve spontaneously whereas in others they persist and may require excision of affected tissue.123,124
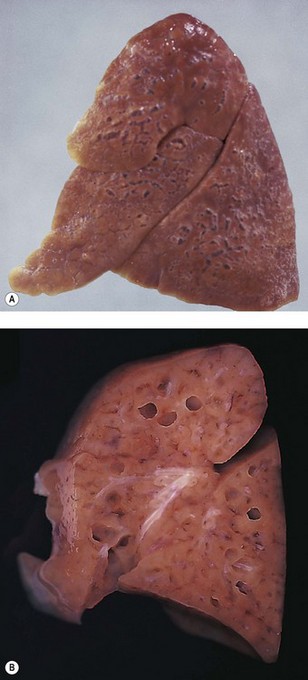
Microscopically the differential diagnosis is from congenital lymphangiectasia (see p. 77). This can be extremely difficult, not least because the air tracks within lymphatics as well as the surrounding connective tissue.125 It is therefore helpful if the nature of the content of the cysts, gaseous or fluid, is ascertained at necropsy. If the emphysema has been present for a few days before death the diagnosis is simplified by the development of a foreign-body giant cell reaction to the air (Fig. 2.18). The presence of interstitial emphysema has been used forensically as evidence of live birth.126
Persistent tachypnoea of infancy associated with neuroendocrine cell hyperplasia
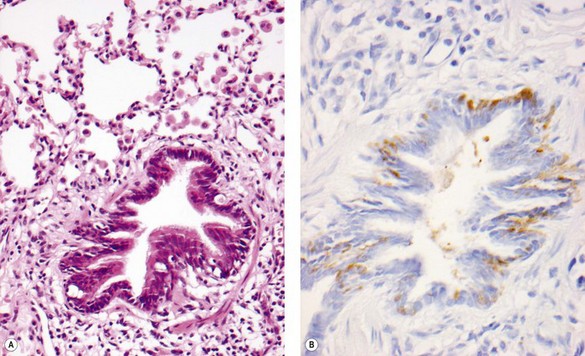
Neuroendocrine cell hyperplasia was identified by immunostaining for bombesin when it was decided to review the archival tissue of a group of infants less than 2 years of age who had presented with persistent tachypnoea, hypoxia, rib retraction or respiratory crackles, and had had a lung biopsy reported as showing seemingly insignificant non-specific changes (Fig. 2.19). These infants showed an increased number of neuroendocrine cells compared to a group of controls.127 The mean onset of symptoms was 3.8 months (range, 0–11 months). Radiographs demonstrated hyperinflation, interstitial markings and ground-glass densities. Oxygen was initially required for prolonged periods, and medication trials did not eliminate symptoms. After a mean of 5 years, no deaths had occurred, and the patients’ condition had improved. It was suggested that these infants represented a distinct group of patients defined by the absence of known lung diseases, the clinical signs and symptoms of interstitial lung disease and idiopathic neuroendocrine cell hyperplasia of infancy. More reports are awaited.
Persistent fetal circulation (persistent pulmonary hypertension of the newborn)
In this condition the high pulmonary blood pressure of the fetus fails to fall at birth and right-to-left shunting through the foramen ovale and ductus arteriosus continues in neonatal life.128 The incidence of the condition is about one in 1000 live births and prior to the introduction of nitric oxide therapy the mortality was about 50%. Autopsy studies show that the muscular pulmonary arteries have a thickened media and that muscle extends into small peripheral arteries that are not normally muscular.129 The condition may be idiopathic or secondary to a wide variety of neonatal cardiopulmonary disorders, including hyaline membrane disease, asphyxia, meconium aspiration and diaphragmatic hernia. The idiopathic cases are poorly understood but autopsy has shown that some are secondary to alveolar capillary dysplasia (see p. 50) that was undetected until autopsy,130,131 whilst others show alveolar dysplasia with good capillary growth.117 All these underlying causes reduce the stimuli that normally initiate pulmonary vascular dilatation at birth, namely pulmonary expansion, raised pulmonary oxygen tension and lowered pulmonary carbon dioxide tension. Notable amongst the agents that mediate the vasodilatation is nitric oxide (endothelium-derived relaxation factor – see p. 22), and the inhalation of low levels of this gas appears to reverse the hypoxaemia of persistent pulmonary hypertension of the newborn.132,133
Diffuse developmental lung disorders
Three conditions come under this heading: congenital acinar dysplasia, congenital alveolar dysplasia and alveolar capillary dysplasia with misalignment of pulmonary veins. The abnormality in each is one of arrested development, the first at the pseudoglandular phase of development, the second at the canalicular or early saccular phase and the third involving the vascularisation of the acinus so that capillaries remain close to their supplying artery in the centre of the acinus and veins also develop in this position rather than in the interlobular septa.117,134
These disorders of development have to be distinguished from several other conditions that affect the neonatal lungs in a diffuse manner. A useful classification of them has recently been promulgated by a North American consortium, dividing them into the diffuse developmental disorders now to be considered, acquired growth abnormalities, specific conditions of undefined etiology and surfactant dysfunction disorders (Table 2.3).134
Table 2.3 Diffuse parenchymal disorders of infancy134
| Disorders | |
|---|---|
| Primary disorders of lung development (aberration in primary molecular mechanism of lung and/or pulmonary vascular development) | 5.8% |
| Congenital acinar dysplasia | |
| Congenital alveolar dysplasia | |
| Alveolar capillary dysplasia with misalignment of pulmonary veins | |
| Growth abnormalities reflecting deficient alveolarisation (usually secondary) | 24.6% |
| Pulmonary hypoplasia | |
| Chronic lung disease of prematurity | |
| Specific conditions of undefined aetiology | |
| Pulmonary interstitial glycogenosis | 3.2% |
| Neuroendocrine cell hyperplasia of infancy | 9.6% |
| Surfactant dysfunction disorders | 9.6% |
| Surfactant protein B mutations | |
| Surfactant protein C mutations | |
| ABCA3 mutations | |
| Histology consistent with surfactant dysfunction but as yet without a recognised genetic etiology: | |
| Alveolar proteinosis | |
| Chronic pneumonitis of infancy | |
| Desquamative interstitial pneumonia | |
| Non-specific interstitial pneumonia | |
| Miscellaneous (e.g. systemic disease processes, immunocompromised states) | 35.5% |
| Unclassified | 11.7% |
Congenital acinar dysplasia
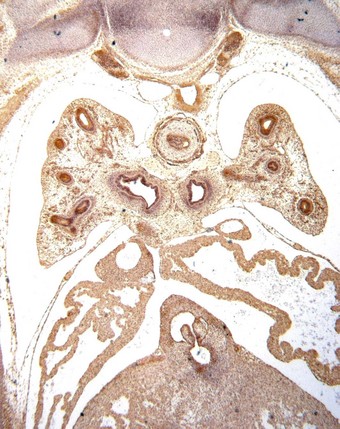
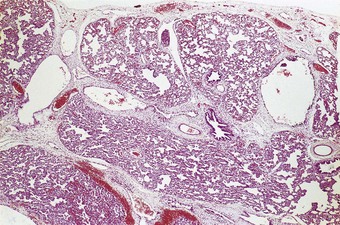
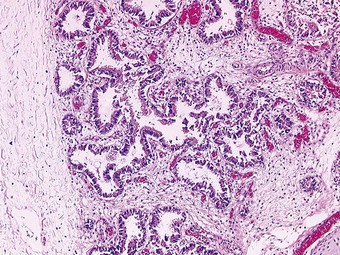
Congenital acinar dysplasia is the rarest and most severe condition of the diffuse developmental lung disorders, being characterised by a complete absence of acinar development. The affected babies, who may be born at term or premature, are cyanosed from birth and survive only a few hours.136–138 They usually have other abnormalities, such as cardiovascular anomalies, dermal hypoplasia and renal hypoplasia. The lungs are small and firm throughout. Microscopically, bronchial-type airways that have cartilage, smooth muscle and glands are separated by abundant mesenchymal tissue. No alveoli are seen (Fig. 2.20).139 An absence of TTF-1, which is implicated in respiratory epithelial differentiation, is reported and occasionally siblings are affected, suggesting that the condition has a genetic basis. It is referred to again on page 59.
Congenital alveolar dysplasia
Congenital alveolar dysplasia was first described in 1948, since when there have been few further reports.140 The condition is characterised by poorly developed air spaces separated by varying degrees of undifferentiated mesenchyme that may histologically resemble pulmonary intersitital glycogenosis. Features of persisitent pulmonary hypertension may also be present. It is generally incompatible with an extrauterine existence but some children survive the neonatal period despite severe respiratory embarrassment.117
Alveolar capillary dysplasia with misalignment of pulmonary veins
Alveolar capillary dysplasia with misalignment of pulmonary veins represents a failure of capillaries and veins to migrate away from the centres of the primitive lung acini. It is an unusual cause of persistent pulmonary hypertension and respiratory distress of the newborn.117,130,141–146 Cases reported to date have all been fatal, with survival generally not exceeding a few hours. Many cases show associated gastrointestinal or genitourinary malformations. Occasionally, siblings are affected and an autosomal-recessive genetic abnormality has been demonstrated in some cases.145,147
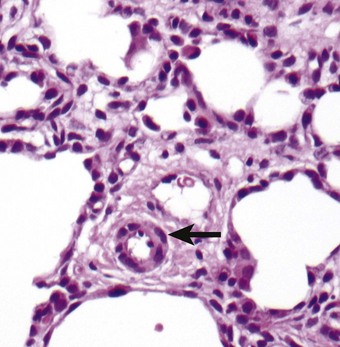
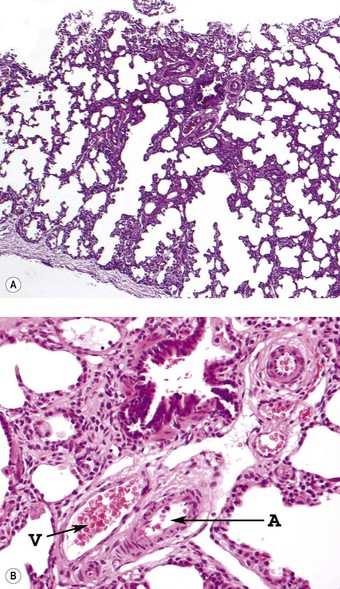
The pulmonary lobules are small and radial alveolar counts are decreased. The alveolar septal connective tissue is increased and alveolar capillaries are greatly reduced (Figs 2.21 and 2.22). Those present are in poor contact with the alveolar epithelium, which shows type II cell hyperplasia.117 The pulmonary veins accompany small pulmonary arteries in the centres of the acini rather than occupying their normal position in the interlobular septa (see Figs 2.21 and 2.22B). The pulmonary arteries are decreased in number and show increased muscularisation. These changes are more easily appreciated if immunostains that localise vascular markers such as CD34 are employed.
Tracheobronchomalacia acquired in infancy
Tracheobronchomalacia (softening of the major airways) may be congenital or acquired but the two are often confused because the condition is most commonly acquired as a complication of congenital lung disease or its treatment, notably intubation.148 This section is concerned with acquired disease. Congenital tracheobronchomalacia is dealt with below. The acquired disease is seen most commonly in premature babies who are intubated because of respiratory distress. Airway infection may also contribute to tracheobronchomalacia being acquired in infancy. Other causes include pressure from congenital abnormalities such as pectus excavatum, bronchogenic cyst or an aberrant artery. Whatever its cause, tracheobronchomalacia causes undue collapsibility of the airways and, hence, respiratory obstruction. Histopathological examination shows fibrous replacement of the tracheobronchial cartilages.
Further disorders of development and growth
Anomalies of the larynx and trachea
Laryngeal atresia
This condition is usually due to cartilaginous overgrowth just below the vocal cords leaving only a narrow channel between the pharynx and trachea that is insufficient to permit respiration. Viewed from above the larynx appears normal but attempts at intubation are unsuccessful and the baby quickly asphyxiates. In utero diagnosis may be possible because the airway obstruction often causes the lungs to be hyperplastic149 and therefore hyperechogenic, in which case an immediate tracheostomy undertaken before the cord is clamped may prove life-saving. However, the condition is often associated with other congenital abnormalities, notably those comprising Fraser’s syndrome. The identification of pulmonary hyperplasia should therefore lead to a search for additional malformations. Pulmonary hyperplasia is described in more detail on page 70.
Tracheal aplasia
With total or partial absence of the trachea, the main bronchi either communicate only with each other, in which case the lungs are hyperplastic, or with the oesophagus.150 A minor degree of tracheal hypoplasia, with both the sagittal and coronal diameters being reduced, may be seen in Down’s syndrome.151 The normal diameters of the adult trachea are given on page 63 under Mounier-Kuhn’s syndrome of tracheobronchomegaly, a genetic disorder which generally becomes evident in adult life.
Tracheal stenosis
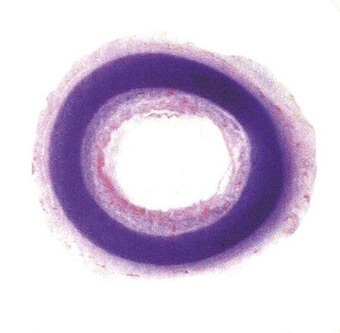
Tracheal stenosis may result from intrinsic or extrinsic lesions, which may be congenital or acquired. Congenital intrinsic stenosis may take the form of a gradual tapering, an isolated segmental narrowing or a membranous web, or be due to a nodule of ectopic oesophageal tissue.154 Congenital extrinsic compression may be due to various anomalies of the great vessels forming (vascular) rings or a sling around the trachea (see p. 481). The left pulmonary artery sling syndrome may form part of a (tracheal) ring–sling complex in which the tracheal cartilages form complete rings, the left bronchus is short and the right is long (bronchial pseudoisomerism).152 The complete cartilage rings represent an absence of the posterior membranous part of the trachea (Fig. 2.23) and the abnormally long trachea is consequently narrowed, the stenosis generally being funnel-shaped. This abnormality is difficult to correct surgically and the prognosis is poor. The trachea normally has 22 cartilages. More are generally seen in the ring–sling complex and fewer in infants with a congenitally short neck. A congenitally short trachea may not be recognised until the seventh decade.153
Tracheo-oesophageal fistula
The mesodermal separation of those parts of the primitive foregut destined to become oesophagus and trachea may be incomplete, resulting in a tracheo-oesophageal fistula (Fig. 2.24). Separation of trachea and oesophagus normally proceeds in a cephalad direction and if this is incomplete the two may communicate. Usually the proximal part of the oesophagus ends in a blind sac and the distal part takes origin from the lower part of the trachea, but the anatomy varies (Fig. 2.25). Histological examination shows that abnormalities in the wall of the trachea extend beyond the fistula; there is often widespread loss of cartilage and squamous metaplasia.154,155 Tracheal communication with remnants of the branchial clefts (branchial cysts) is also recorded.156 Occasionally it is a bronchus that communicates with the oesophagus.157 Tracheo-oesophageal fistula sometimes forms part of a so-called VACTERL association, which represents a non-random grouping of birth defects involving mesodermal structures for which no genetic basis has yet been identified, VACTERL being a mnemonic for vertebral, anal, cardiovascular, tracheoesophageal, renal and limb abnormalities.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


