Chapter 3 Diseases of the conductive airways
The function of the airways is to conduct gas in and out of the lungs and all airway diseases are liable to impede this, resulting in ‘obstructive lung disease’, as opposed to the ‘restrictive lung disease’ that is caused by many diseases of the lung parenchyma. Airway obstruction has important effects on the lung parenchyma and this chapter first considers one of these, pulmonary collapse. Another important consequence of airway obstruction is obstructive pneumonia, which is dealt with on page 315, while if airway obstruction is not relieved, distal infection is almost inevitable.
Atelectasis and pulmonary collapse
Absorption collapse is likely when bronchial obstruction prevents free entry of air into the lungs. The causes are listed in Box 3.1. Mucus frequently collects during anaesthesia, when respiratory movements are reduced and the cough reflex suppressed, whilst mucus plugging is a cardinal feature of asthma, allergic bronchopulmonary aspergillosis and plastic bronchitis. The inhalation of a foreign body is especially common in children while the narrow, pliable bronchi of infants are particularly liable to be compressed by distended pulmonary arteries at points where they are in close anatomical proximity (see Fig. 10.9, p. 481) or by abnormally located systemic arteries. In time, the alveolar air – first the oxygen and later the nitrogen – is removed by the blood that passes through the affected area and the alveoli then progressively collapse.
Box 3.1 Causes of absorption collapse
aTumours particularly prone to grow into the lumen of an airway include carcinosarcoma, carcinoid, bronchial gland neoplasms, metastases, lymphoma, chondroid hamartoma, papillary neoplasms, granular cell tumour, amyloid tumour and broncholith.
Pulmonary collapse has been seen quite commonly in the crew of high-performance aircraft. An important cause is breathing pure oxygen, which washes nitrogen from the alveoli and is more rapidly absorbed into the blood. Parts of the lung filled with oxygen but temporarily closed off by increased gravitational forces distorting airways are liable to absorption collapse. These forces operate whenever the pilot makes a tight turn at high speed or pulls out of a steep dive. Clothing designed to protect the aviator from burst lung (see p. 369) increases the adverse effect on the basal parts of the lungs by raising the diaphragm and reducing lung volume.
Pathological findings
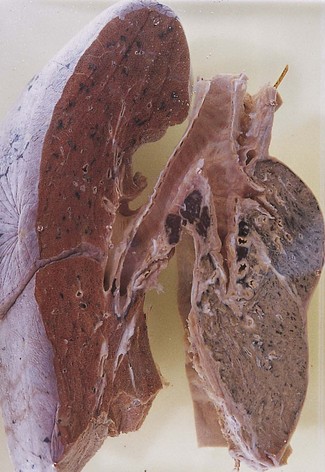
Whatever the cause, collapsed lungs are small and firm, and have a deeply wrinkled pleural surface. Portions of collapsed lungs tend to sink when dropped into water but this is not an infallible test of airlessness. Where part of a lung has recently collapsed, the immediately adjacent, pale pink, aerated lobules are sharply separated from the dark red depressed areas of collapse by zigzag lines that correspond to the interlobular septa (see Fig. 4.3, p. 137). The collapse involves alveoli and bronchioles but bronchial cartilage maintains the patency of these larger airways. Subsequent changes differ according to whether the collapse is due to absorption or compression. With absorption collapse the affected lung resembles splenic tissue, both grossly and microscopically.1 Alveolar walls are in apposition and their capillaries are greatly dilated so that the bulk of the collapsed lung no longer consists of air space but of sinusoidal vessels engorged with blood, although the circulation may be reduced. The interlobular septa are thickened but there is little fibrosis of the alveolar tissue. The changes are irreversible, suggesting that there is fusion of the apposed alveolar walls. With pressure collapse congestion is less marked and fibrosis of both the alveolar tissue and overlying pleura is more marked (Fig. 3.1). In either case, reinflation is prevented.
Collateral ventilation
When obstruction to an airway is only partial, permitting inspiration but hindering expiration, air is retained in the affected area and there is full inflation rather than collapse (see infantile lobar emphysema, p. 72). Absorption collapse may also be prevented by collateral ventilation, a process by which one portion of lung is ventilated through another via the pores of Kohn and other peripheral communications (see p. 15). Collateral ventilation is best developed at the acinar level, being hindered by the interlobular septa and prevented by interlobar fissures, but because interlobular septa are incomplete, it may prevent whole segments undergoing absorption collapse. However, air flow through the tortuous bypass channels afforded by many pores of Kohn is poor and collateral ventilation plays little part in gas exchange.2 Its function is to maintain inflation of alveoli when their supplying airways are obstructed by secretions or a foreign body. This is essential to the cough mechanism, dependent upon which is the expulsion of the obstructive material and the restoration of bronchial patency.
Middle-lobe syndrome
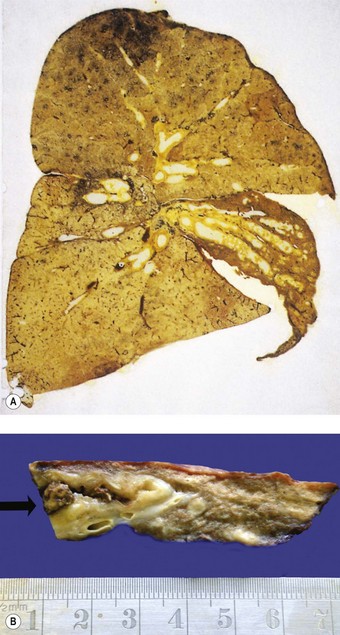
The term ‘middle-lobe syndrome’ was coined to describe a condition of chronic or recurrent absorption collapse of the right middle lobe that was originally described in 1937.3 The collapse was most frequently caused by tuberculous involvement of lymph nodes compressing the right middle-lobe bronchus. Tuberculosis is less common today but any disease enlarging these lymph nodes may have the same effect. Predilection for involvement of the middle lobe was considered to be the result of a combination of factors: the prominent collar of nodes about its bronchus, the lymphatic drainage of these nodes being from much of the right lung and parts of the left, and the relatively narrow calibre and possibly undue compressibility of the middle-lobe bronchus. A further possible factor is the limited capacity for collateral ventilation (see above) within the middle lobe: this stems from the fact that its two segments have relatively large proportions of their surfaces covered by pleura and, together with the inferior segment of the lingula, are the only ones that abut no more than one other segment.4
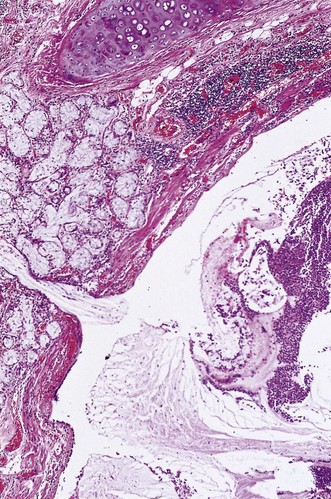
Patients with the middle-lobe syndrome complain of chronic cough, haemoptysis, chest pain and dyspnoea, and the diseased lobe may be removed to relieve this. Pathological changes in the resected lobe include bronchiectasis, chronic bronchitis and bronchiolitis, lymphoid hyperplasia, organising pneumonia and abscess formation, in addition to collapse (Fig. 3.2).5 A similar syndrome may affect the lingula.
Bronchiectasis involving the middle lobe or lingula of middle-aged women has also been termed the Lady Windermere syndrome, based upon the belief that such a fastidious person as the eponymous person in Oscar Wilde’s play Lady Windermere’s Fan would refrain from expectorating and thereby retain any infected bronchial secretions.6 However, the bronchi to these portions of the lungs extend forwards so that unless the person spent a considerable time in the prone position the middle lobe and lingula are the least likely parts of the lungs to be affected by aspiration (see Fig. 5.2.14, p. 189).7 Infection (or colonisation) by Mycobacterium avium-intracellulare is frequently identified in this ‘syndrome’.
Obstruction of the upper airways
Obstruction of the upper airways may be complete, and cause rapid asphyxial death, or incomplete, when there is stridor or wheezing, or distal complications such as obstructive pneumonia may ensue. Foreign bodies are an important cause, especially in children and edentulous adults. Tumours are another important cause. Rare causes include amyloid tumours, Wegener’s granulomatosis8 and tracheobronchomalacia. In infancy the airways are unduly pliable and may be compressed by distended arteries, particularly where the two are in close contact (see Fig. 10.9, p. 481). Anomalous arteries may also compress airways in infancy, as in the vascular sling and ring syndromes (see Figs 10.10 and 10.11, pp. 481, 482).
Obstructive sleep apnoea
Obstructive sleep apnoea is characterised by repeated periods during which the patient stops breathing for 10 or more seconds whilst asleep. The patient may not waken but is repeatedly aroused so that the quality of the sleep is poor and daytime sleepiness is consequently excessive.9 Snoring is a common accompaniment. The patient is generally obese and it is postulated that cervical fat pads obstruct the upper airway.10 Other family members are often similarly affected, possibly because of similarities in cervicofacial structure. Charles Dickens was evidently familiar with such individuals, portraying one in his novel The Pickwick Papers. Such patients have therefore been termed ‘pickwickian’, although the Dickens character was the ‘fat boy’ rather than Mr Pickwick himself. Defects in the secretion of testosterone and growth hormone may also be identified.11 These are reversible and are probably due to the central effects of sleep fragmentation and hypoxaemia.
The principal problem in obstructive sleep apnoea is the daytime tiredness, which leads to poor performance at work and a tendency to fall asleep at inappropriate moments. The consequences of this can be very serious if, for example, the patient drives. The periods of apnoea result in hypoxaemia, which in turn causes pulmonary hypertension. The apnoeic episodes are also accompanied by systemic hypertension and death may be caused by biventricular cardiac failure.12 The pulmonary blood vessels show the usual changes found with hypoxia, principally hypertrophy of the arterial media (see p. 424). Pulmonary haemorrhage and haemosiderosis are further features, possibly attributable to the left ventricular failure. Pronounced capillary proliferation resembling capillary haemangiomatosis (see p. 429) is also described.13 Continuous positive airway pressure provides effective treatment.14
Obstructive sleep apnoea is to be distinguished from a central variety of apnoea known as Ondine’s curse. In German legend, the water nymph Ondine, having been jilted by her mortal lover, took from him all automatic functions, requiring him to remember to breathe. When he finally fell asleep, he died. Central apnoea has been encountered with bulbar poliomyelitis and it is likely that the syndrome results from damage to the medullary CO2 receptor. Airway patency is maintained but respiratory drive is weak, especially during sleep.
Tracheobronchopathia osteochondroplastica
The first descriptions of this condition date back to the middle of the nineteenth century,15,16 since when it has continued to arouse interest because of its apparent rarity and disputed aetiology.17,18 It is confined to the trachea and bronchi and does not infiltrate surrounding tissues or metastasise but it endangers life through airway obstruction.19 It affects men more often than women, and is seldom recognised before the age of 50. Symptomatic cases are rare but it is possible that mild cases are overlooked20: four cases were reported in one series of 500 bronchoscopies.21
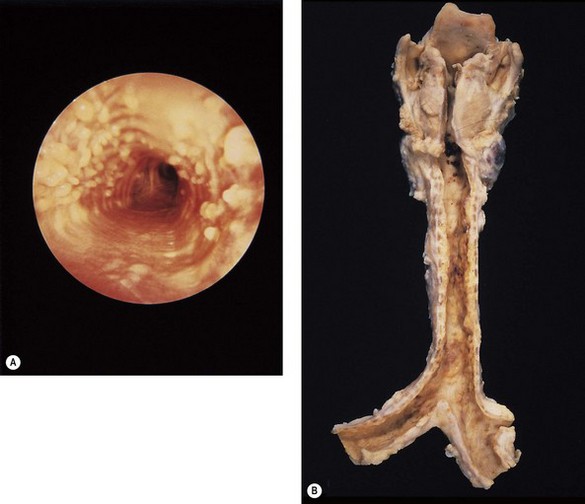
Tracheobronchoscopy reveals multiple mucosal nodules and relevant to both the diagnosis and aetiology of the condition is the observation that the membranous portion of the trachea is spared (Fig. 3.3).22 This suggests that the condition is related to the airway cartilage and that the lesions represent exostoses (as suggested by Virchow) rather than submucosal metaplasia (as suggested by Aschoff).16,23 A superficial resemblance to tracheobronchial amyloidosis (which is prone to ossify) has led to erroneous suggestions that these two conditions are related.24–26 Growth factors that induce new bone formation have been demonstrated about the ossifying nodules but not about those composed of mature lamellar bone.27
Pathology
At necropsy the tracheobronchial mucosa is roughened by numerous nodular excresences (see Fig. 3.3B). Microscopy shows that the nodules consist of cartilage, which, like the normal cartilage of the airways may calcify and ossify.19,22,28–30 These osseocartilaginous nodules are situated between the normal cartilage and the surface epithelium of the airway, causing the mucosa to protrude into and compromise the lumen. The new cartilage differs from that normally found in the airways only in its abnormal position. Cytologically it is quite normal and in a small fibreoptic biopsy is likely to be mistaken for the normal cartilage of the large airways. It generally appears to have no connection with the normal cartilage but step sections show that there is indeed continuity through narrow pedicles,22,30 supporting the view that the condition represents multiple ecchondroses of the tracheobronchial cartilages,28 as originally proposed by Virchow.16 Treatment consists of nibbling the nodules away endoscopically as often as proves necessary.
Relapsing polychondritis
This condition is characterised by recurrent inflammation of cartilaginous structures and other tissues rich in glycosaminogycans.31–35 Immunoglobulins and complement have been identified at the chondrofibrous junction,36 and the presence of circulating anticartilage immunoglobulin and the ability of cartilage antigens to transform lymphocytes from these patients provide evidence that the disease has a tissue-specific autoimmune basis.37
Clinical features
The disease affects patients of both sexes and any age but the maximum frequency is in the fourth decade. It typically causes distortion of the pinnae and collapse of the nose. Other tissues involved include the larynx, trachea, bronchi, joints, eyes, inner ears and blood vessels. The trachea and bronchi are involved in 21% of patients but very rarely are they affected in isolation.38–42 Tracheobronchial involvement is characterised by airflow obstruction due to airway collapse, manifest as breathlessness, cough, hoarseness and stridor.38–40 Less commonly there is bronchorrhoea.43 The arthritis has a predilection for thoracic joints and may further contribute to respiratory difficulties. Blood vessel involvement is characterised by vasculitis involving vessels of all sizes and leading to aneurysms of major arteries. Occasionally, medium-sized arteries develop aneurysms and the changes are then those of polyarteritis nodosa.44 Glomerulonephritis may also develop.45
Pathology
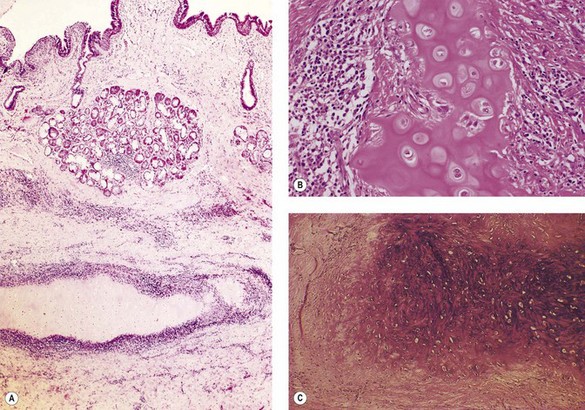
The affected bronchi may feel soft. Microscopically, the appearances vary according to the degree of inflammatory activity. In the active stage of the disease, the tracheobronchial cartilage is less basophilic than normal, reflecting loss of acidic proteoglycans, which may appear in the urine.33 The tracheal and bronchial cartilages are cuffed by a chronic inflammatory infiltrate of lymphocytes, plasma cells and occasional multinucleate histiocytes that is limited to the edge of the cartilage, which is ragged and evidently under attack (Fig. 3.4A, B).32 In the late stages of the disease the inflammation may have resolved, leaving collagen surrounding and intersecting the cartilage matrix, which at this stage is fibrillary rather than amorphous and shows increased basophilia (Fig. 3.4C).33,38,40 Other components of the airway appear normal and there is generally no evidence of vasculitis in the airways. These features are characteristic but not specific, being seen for example in a postintubation tracheal stricture.
Idiopathic tracheal stenosis
Idiopathic tracheal stenosis is reputedly rare but one group was able to review 63 such patients who had required surgery at one hospital over a 19-year period.46 The patients were all middle-aged women (mean age 49 years), one-third of whom gave a history of gastro-oesophageal reflux. None had evidence of rheumatoid disease, systemic lupus erythematosus or polychondritis and ANCA testing was uniformly negative. The stenosis affected the subglottis or upper one-third of the trachea and showed extensive keloid formation with dilatation of the tracheal glands. A form of fibromatosis was postulated.
Acute tracheobronchitis and bronchiolitis
Environmental causes
In men engaged in industries in which irritant gases or dusts may be inhaled, the mucous membrane of the trachea and bronchi may become acutely inflamed, and occasionally noxious gases such as ammonia, oxides of nitrogen and sulphur dioxide may be breathed in such concentrations that widespread injury to the respiratory mucosa may follow. Silo-filler’s disease, described on page 356 [ch 7.1], is one such example. The use of thermal lances on steel is ordinarily safe but if special alloys of steel are attacked with these tools the inhalation of beryllium, cadmium and other hot metal fumes may cause acute bronchiolitis and diffuse alveolar damage. In the First World War, the military use of chlorine and phosgene as poisonous gases was often followed by destructive lesions throughout the respiratory tracts of the exposed troops.
The damage inflicted by soluble noxious gases and fumes is liable to be concentrated on the main airways whereas less soluble gases are prone to damage more distal air spaces, including alveoli as well as the finer conductive airways (see Fig. 7.2.2 and Table 7.2.2, p. 372).47 Examples of the former include chlorine and ozone, whilst the latter include beryllium, mercury and cadmium fumes, oxides of nitrogen and high concentrations of oxygen.
Microbial causes
In recent decades, great changes have taken place in the relative importance of bacteria and viruses in the aetiology of acute tracheobronchitis.48 Prophylactic immunisation against measles, diphtheria and pertussis, and the availability of antibiotics effective against the bacterial causes of secondary pneumonia, particularly pneumococci, have together greatly lessened the frequency of both the primary diseases and the respiratory complications. Most of these microbial diseases are dealt with in the chapters devoted to viral and bacterial infections (Chapters 5.1–5.3) but diphtheria and whooping cough will now be described.
Whooping cough (pertussis)
At one time, whooping cough was one of the commonest causes of death in children, and its decline in the developed countries of the world since the Second World War represents one of the notable contributions of prophylaxis to public health. In the UK, for example, widespread immunisation resulted in a 30-fold reduction in the number of notifications, and deaths became rare. Understandably, complacency followed, and when publicity was given to cerebral complications of the vaccine in 1974, its acceptance rate dropped dramatically, followed in 1977 by the biggest epidemic for 20 years (Fig. 3.5). Subsequent studies showed that the risk of permanent brain damage was very small – 1 in 310 000 injections. Increased vaccine uptake resulting from a return of public confidence cut short an expected epidemic in 1986 and in 1991, when uptake had risen to 88%, an anticipated epidemic failed to materialise.
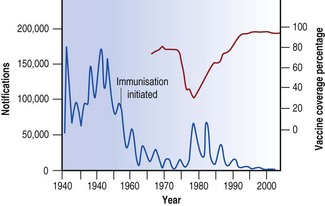
B. pertussis has a marked tendency to attach itself to respiratory epithelium.49 In fatal cases, B. pertussis can be recovered from the lungs, and the organisms can be seen microscopically in large numbers in the thick mucopurulent film that covers the mucosa of the trachea and the bronchi. The mucus may be so viscous that it obstructs the passage of air and so leads to segmental lung collapse. Spread of the infection via the airways results in bronchopneumonia. In recent years intractable pulmonary hypertension has been recognised in some fatal cases, with large collections of leukocytes being found within the pulmonary vessels in such cases.50
Necrotising sialometaplasia
Throughout the lower respiratory tract regenerative processes may be so atypical that carcinomatous transformation has to be considered in the differential diagnosis. This impression is often augmented by excessive mitotic activity and metaplasia. Thus, at the alveolar level necrotising lesions such as infarcts and the granulomatoses may be bordered by foci of atypical squamous hyperplasia that are easily mistaken for squamous cell carcinoma. Similarly, damage to the bronchial epithelium is often followed by atypical regeneration that is easily mistaken for carcinoma,51 particularly when exfoliated cells are being examined. Bronchoscopy inevitably involves bronchial injury and cytopathologists have to be aware of the atypicalities that follow this procedure. Necrotising lesions of the larynx are sometimes accompanied by atypical regeneration that involves both the surface epithelium and the submucosal glands: the term ‘necrotising sialometaplasia’52,53 has been applied to this and to a similar process involving the trachea in patients with herpetic tracheitis undergoing repeated intubation.54,55 Microscopically there is coagulative necrosis of the tracheal glands with the ghost outlines of the acinar cells retaining a lobular distribution. There is often mucous extravasation and a mixed acute and chronic inflammatory infiltrate. Regenerative hyperplasia, often with squamous metaplasia, is usually first evident in the gland ducts. The changes are benign and self-limiting but in small biopsies may be mistaken for those of squamous cell carcinoma or mucoepidermoid carcinoma. Identification of the investing myoepithelial cells by staining for smooth-muscle actin favours sialometaplasia as these cells are soon destroyed by invasive growths.56 Low immunoreactivity for p53 also favours this benign process.53
Chronic obstructive pulmonary disease57
Morbidity and mortality
COPD is the fourth leading cause of death in Europe and North America and with the increase in smoking in developing countries it is expected to become the third leading cause of death worldwide by 2020.58 Death comes many years after the onset of the disease and COPD is therefore also a major cause of sickness and incapacity for work. The social gradient of the disease is steep; the death rate in the poorest section of the population is some five times that in the most prosperous. Death is due to respiratory insufficiency, often complicated by bronchopneumonia, cardiac failure or pulmonary thromboembolism.59 There is also a four- to fivefold increased risk of lung cancer in patients with COPD, as compared with controls matched for cigarette smoking.60,61
Clinical features
Patients with COPD are generally current or former smokers over 35 years of age. The sex ratio reflects recent smoking patterns so that in the UK, for example, the disease formerly showed a marked male predominance but is now becoming less common in men and more common in women. Whether or not there is a sex difference in susceptibility is uncertain.61a
Patients complain of breathlessness, impaired exercise tolerance and cough. Wheeze is more suggestive of asthma but the conditions may coexist. The cough is particularly likely to be productive in chronic bronchitis. The sputum is typically mucoid and white but the disease is marked by episodes of acute bronchitis when the sputum becomes purulent and yellow. Later the sputum may become purulent continuously; it accumulates in the bronchi during sleep and causes severe obstruction of the airways until it is coughed up in the morning. Whilst a change from white to yellow sputum usually signifies infection it should be noted that large numbers of eosinophils also render the sputum yellow, a potential pitfall in the clinical distinction of bronchitis and asthma.
Microbiological examination of the sputum in chronic bronchitis has shown that the most frequent and important pathogens are Haemophilus influenzae, Streptococcus pneumoniae, Branhamella (Moraxella) catarrhalis and Chlamydia pneumoniae.62–67 Purulent sputum usually contains one or more of these organisms in abundance: they tend to disappear after antimicrobial therapy when the sputum becomes mucoid again.
The productive cough appears at first only in the winter months; later it is present all through the year, characteristically with acute exacerbations in winter that are usually precipitated by a viral infection.68,69
The majority of patients with COPD suffer from both obstructive airway disease and emphysema but a minority of patients have only one of these conditions. Two clinical syndromes, types A (‘pink puffer’) and B (‘blue bloater’), have been described with the former thought to indicate emphysema and the latter chronic bronchitis.70,71 The association of the type A syndrome with emphysema is fairly well established but the association of type B with chronic bronchitis is not well substantiated morphologically. Type A patients show rapid shallow breathing and this maintains near normal blood gases at the cost of subjective breathlessness. They are usually thin and because their blood gases are not severely deranged they tend not to develop polycythaemia or cor pulmonale. Type B patients on the other hand are hypoxic and therefore suffer from polycythaemia and repeated bouts of congestive cardiac failure. They are usually obese and oedematous and have a productive cough but they are seldom severely breathless. It is important to realize that most patients with chronic airflow limitation do not fit neatly into one or other of these types. Nor do these two types reflect pure bronchitis or pure emphysema.72 The fundamental difference between type A and type B patients may be in the brain rather than the lungs: type B patients seem to have a respiratory centre that is relatively unresponsive to the usual stimuli, an abnormality that may be genetically determined.
Chronic bronchitis
Definition
Chronic bronchitis is defined in clinical terms as a persistent or recurrent excess of secretion in the bronchial tree on most days for at least 3 months in the year, over at least 2 years.73 The secretions of the normal human respiratory tract are believed to total less than 100 ml in 24 hours, all of which is swallowed without the conscious need to clear the throat or cough so that the normal person produces no sputum. The diagnosis of chronic bronchitis may be made only when other conditions that cause expectoration, such as tuberculosis and bronchiectasis, have been excluded.
Chronic bronchitis was formerly subdivided into simple mucoid bronchitis, mucopurulent bronchitis and obstructive bronchitis.74 It was widely thought that these subdivisions represented successive phases of the disease but a strong counterargument to this was advanced by British epidemiologists.75 These workers showed that, while simple mucoid bronchitis progresses to the mucopurulent variety, this does not progress in turn to the obstructive form. In line with this, neither bronchial gland size nor sputum production is significantly related to airflow limitation.76 The view that the development of obstructive bronchitis is independent of the repeated respiratory infections that characterise mucopurulent bronchitis has been challenged77 but the obstructive form of the disease is now nevertheless widely recognised as a separate condition – small-airway disease.
Aetiology
Chronic bronchitis affects mainly the middle-aged and elderly and is commoner in men; cigarette smoking is by far the most important cause.58,78,79 The influence of cigarette smoke often begins in infancy when the child is exposed passively to parental cigarette smoke. This is generally augmented by active cigarette smoking when the child emulates parents or schoolmates and acquires the habit, often becoming addicted for life. However, as with lung cancer, many indulge in smoking with impunity, indicating that susceptibility to disease varies considerably,57 probably reflecting genetic differences in the control of factors such as the balance of helper and cytotoxic T lymphocytes.80 Marijuana smoke is likely to be recognised as a further aetiological agent as it has similar morphological effects on the airways as tobacco smoke.81
Other factors contributing to chronic bronchitis include general air pollution, which accounts for the higher prevalence of the disease in urban communities, domestic air pollution, which is important in many poor communities due to the combustion of biomass,82–84a occupational dust exposure,85,86 fog, and a damp, cold climate. The morbidity from the disease rises every winter, and remains high throughout the colder, damper months. The occurrence of fog, especially that form known as smog in which the water vapour becomes heavily contaminated with smoke and sulphurous gases, causes a prompt increase in both morbidity and mortality among older people. The heavy 4-day smog in London in 1952 is believed to have precipitated 4000 deaths.
Infections by respiratory viruses and bacteria are also of importance in both initiating and promoting chronic bronchitis.87 Some patients may recall a liability in their earlier years for head colds to go to their chest.88 Relatives are often similarly affected. An increased frequency of respiratory infection in childhood has been identified in adults with chronic bronchitis.89
Morbid anatomy
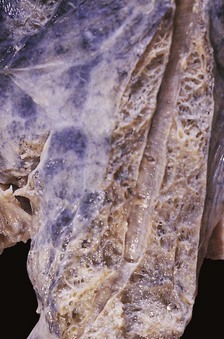
When the lungs of a patient with chronic bronchitis are dissected at necropsy, the exposed bronchi, especially those in the lower lobes, are typically filled with a mixture of mucus and pus. When the purulent material is washed away from bronchi that have been opened longitudinally, the underlying mucous membrane is seen to be a dusky red. The calibre of the main bronchi may remain unchanged but distal bronchi characteristically are slightly dilated: when they are opened with fine scissors, the dilation is found to reach almost to the pleura (Fig. 3.6).90 Some consider the dilation to be due to atrophy of the bronchial wall and describe it in association with emphysema rather than as a feature of chronic bronchitis.91,92 Others have described degenerative changes in the bronchial cartilage in chronic bronchitis and emphysema and have correlated this with the degree of inflammation.93 The lung substance is often emphysematous in patients with chronic bronchitis and there may be bronchopneumonia.
Histological appearances
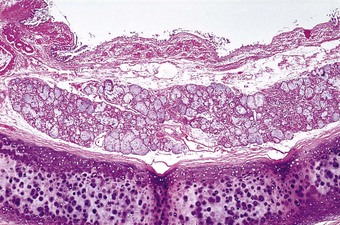
The main features of chronic bronchitis become apparent only when the lungs are examined histologically. The submucosal glands are much enlarged, and there is a shift in gland type from mixed seromucous to pure mucous (Figs 3.7 and 3.8).94 The enlargement is primarily a hyperplastic change.95 Furthermore the usual mixture of neutral and acidic glycoprotein in bronchial mucus changes to one that is largely acidic and within the acidic mucins sulphomucin increases at the expense of sialomucin, alterations that possibly increase sputum viscosity. The mucous acini and their ducts become distended with retained mucus.94,96
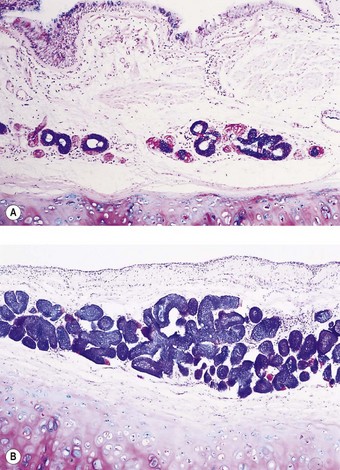
It is possible to correlate the clinical history of chronic bronchitis with the size of the bronchial glands. This may be done by measuring the ratio of the thickness of the gland layer to the thickness of the wall between the base of the surface epithelium and the internal limit of the cartilage plates. The fraction occupied by the glands is known as the Reid index.94 In chronic bronchitis this may double from the normal value of 0.3 (see Figs 3.7 and 3.8). The Reid index takes no account of the glands situated between the cartilaginous plates and a more accurate method of assessing the size of the glands is to estimate their cross-sectional area as a percentage of that of all the bronchial wall components.97,98 This is now greatly facilitated by the use of a computerised digitising tablet rather than relying on the accurate but tedious method of point counting.
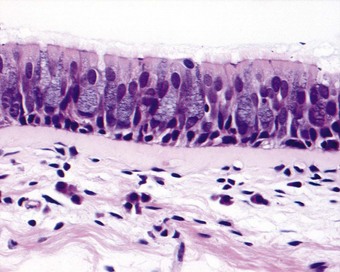
As well as the glands, the epithelium that lines the bronchi shows signs of increased mucus production, the proportion of goblet cells being increased at the expense of the ciliated cells (Fig. 3.9).99 Also, the surface epithelium may undergo patchy squamous metaplasia. The accompanying loss of cilia, which ordinarily clear bacteria and dust particles from the lower respiratory tract, predisposes to infection.
The fall in the proportion of serous to mucous acini in the bronchial glands may also be expected to promote infection as the serous cells are a major source of the antibacterial agents lysozyme and lactoferrin100,101; however, these antibacterial agents continue to be detectable in patients’ sputum.102 The serous cells also contribute the secretory piece to immunoglobulin A, so protecting it from proteolytic degradation. Reduced expression of this secretory piece has been demonstrated in severe COPD.103 The serous cells are also a major source of secretory leukocyte protease inhibitor.104 This factor can be identified in the sputum of patients with chronic bronchitis but normal values have not been established: a diminution could help explain why chronic bronchitis and emphysema are so commonly associated.
Mucus accumulates in the airways and may completely fill their lumen. With infection, neutrophil leukocytes are added to the mucus and the airway wall is swollen by oedema and an acute inflammatory infiltrate; between acute attacks, the wall of the airways is infiltrated by lymphocytes and macrophages and its blood vessels are congested. The lymphocytes are largely CD8-positive (cytotoxic/suppressor) T cells, in contrast to the CD4-positive Th2 cells found in asthma.99,105,106 Thus, although essentially hypersecretory, chronic bronchitis is also a truly inflammatory disease (Fig. 3.10).107 Furthermore, it appears that the inflammation in turn promotes hypersecretion,108,109 a process envisaged by an older generation of pathologists when they spoke of catarrhal inflammation. A vicious cycle is thus set in motion in chronic bronchitis, as in bronchiectasis (see p. 119). In contrast to bronchiectasis, however, the musculature and cartilage of the bronchial walls remain intact in chronic bronchitis.90
‘Wheezy’ bronchitis
Hyperplasia of bronchial muscle in chronic bronchitis is reported by some observers110 but not by others.111 The explanation for this discrepancy may be that some patients have features of both asthma and bronchitis. The amount of muscle in the airways of these ‘wheezy bronchitics’ is intermediate between the normal amounts found in purely bronchitic subjects and the increased amount found in atopic asthmatics.112
Constitutional factors, in particular bronchial hyperactivity, may contribute to airflow limitation in certain cigarette smokers who develop ‘chronic asthmatic bronchitis’.113 This concept is embodied in what has become known as the ‘Dutch hypothesis’ – that smokers with progressive airflow limitation have increased bronchial reactivity and atopic features similar to, but less marked than, those observed in asthma.114 The observation that cigarette smokers have elevated serum immunoglobulin E levels raises the possibility that some of the adverse effects of smoking might be immunologically mediated.115 However the elevated serum immunoglobulin E in smokers does not appear to be specific for the common seasonal aeroallergens. Chronic bronchitis and asthma are compared in Table 3.1.116
Table 3.1 Comparison of chronic bronchitis and asthma116
| Chronic bronchitis | Asthma | |
|---|---|---|
| Age | Over 35 years | Any |
| Smoking history | Almost invariable | Possible |
| Cough | Persistent and productive | Intermittent and non-productive |
| Breathlessness | Progressive and persistent | Intermittent and variable |
| Nocturnal symptoms | Uncommon | Common |
| Family history | Uncommon unless family members also smoke | Common |
| Concomitant eczema or allergic rhinitis | Uncommon | Common |
| Airflow obstruction | Fixed and irreversible | Variable and reversible |
| Sputum | Macrophages Neutrophils | Eosinophils Charcot–Leyden crystals Creola bodies Curschmann’s spirals |
| Postmortem appearances | Excess mucus Bronchial dilatation Associated emphysema | Mucous plugs Hyperinflation but no emphysema |
| Airway inflammation | CD8+ T cells Neutrophils periodically | CD4+ T cells Eosinophils Mast cells |
| Airway congestion and oedema | Present | Present |
| Airway epithelium | Intact Goblet cell hyperplasia Squamous metaplasia | Fragile with stripping Goblet cell hyperplasia Squamous metaplasia |
| Basement membrane reticular layer thickening | Mild-to-moderate | Marked |
| Bronchial glands | Marked enlargement of the mucous acini | Moderate enlargement of both mucous and serous acini |
| Mucin histochemistry | Shift from neutral to acid and within the acid mucins from sialo- to sulpho- | Unchanged |
| Airway muscle | May show hypertrophy | Marked hypertrophy |
| Major complications | Cor pulmonale | Allergic bronchopulmonary aspergillosis |
Small-airway disease (chronic obstructive bronchiolitis)
The aetiological differences between chronic bronchitis and small-airway disease are as yet unclear but cigarette smoking is undoubtedly important in both. The latter condition is met more often in those patients whose breathlessness steadily increases with the years and in whom there is progressive deterioration in exercise tolerance leading to inability to continue working.
Postmortem studies have shown that in chronic obstructive lung disease the major site of airflow obstruction is in airways of about 2 mm diameter or less.117 Airways of this calibre, which correspond to those of approximately the eighth generation, have subsequently become generally known as ‘small airways’. They include both small bronchi and proximal bronchioles.
Such small-calibre airways are numerous and in health they have a large collective cross-sectional area so that they normally contribute little to total airflow resistance (see Fig. 1.8, p. 7). Many may be lost before there is any appreciable impairment of airflow. It is likely therefore that many cigarette smokers are progressively developing obstructive airway disease long before they notice any significant reduction in their respiratory capabilities. For this reason the periphery of the lung has become known as its ‘silent zone’.
Histopathology
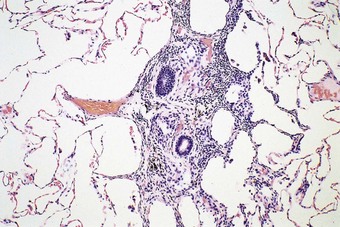
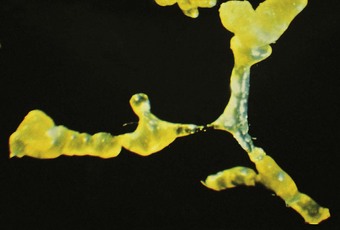
Small-airway disease is characterised by bronchiolar goblet cell hyperplasia.118 This takes place at the expense of Clara cells,119 which, together with the serous cells of the bronchial glands, secrete an airway-specific low-molecular-weight protease inhibitor (antileukoprotease), which is a potent protective factor against the development of emphysema.104,120–123 There is also inflammation in the smaller bronchi and bronchioles. Similar chronic inflammatory changes to those affecting the larger airways in chronic bronchitis are observed in the walls and adjacent tissues of bronchioles and small bronchi, the predominant cell again being the CD8-positive T lymphocyte.124 Wall thickening125 and fibrosing peribronchiolitis126 (Fig. 3.11) lead to the lumen becoming severely reduced: this causes irreversible obstruction and severe airflow limitation. The narrowing takes the form of focal stenoses.127 Proximal to the stenoses the bronchioles are often dilated. Bronchographic medium pools in the dilated segments, giving what has been described as a ‘mimosa flower’ effect,128 and there is an absence of peripheral filling.129 The focal stenoses are difficult to identify in random sections but are well demonstrated in plastic casts of the airways (Fig. 3.12).130,131 Alternatively, quantitative methods may be employed: these show both organic narrowing and mucus plugging of small airways.132 It is likely that cases of small-airway disease were included among the patients with chronic lung diseases studied by McLean.133–135 In many smokers peribronchiolar inflammation and fibrosis involve the more distal respiratory bronchioles and thicken the walls of adjacent alveoli so that there is restrictive as well as obstructive lung disease. This so-called respiratory bronchiolitis-associated interstitial lung disease overlaps with yet another effect of cigarette smoking, namely desquamative interstitial pneumonia, and is dealt with on page 313.
Complications
Patients with small-airway disease are prone to develop cor pulmonale, mainly as a result of widespread hypoxic pulmonary vasoconstriction and the consequent rise in pulmonary vascular resistance. Hypoxic pulmonary hypertension is dealt with on page 424. A further consequence of the hypoxia is a compensatory polycythaemia, the resultant haemoconcentration adding to the increased cardiac burden. Whilst death from small-airway disease is usually due to right-sided heart failure, obstructive respiratory failure and bronchopneumonia also contribute. These conditions are often present in combination.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


