Chapter 15 Neutrophilic and eosinophilic dermatoses
Pyoderma gangrenosum
Clinical features
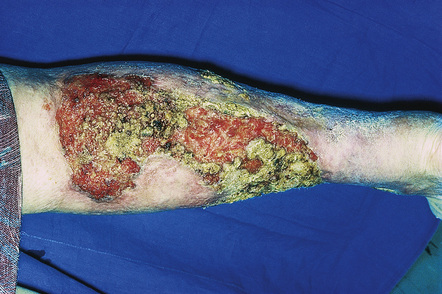
Pyoderma gangrenosum is an uncommon disease of obscure etiology.1–11 It appears to be somewhat more common in women and, although it may occur at any age, most patients are in their fourth or fifth decade.4 Presentation in children is uncommon, but it has been seen even in infants,12–21 and rare familial cases have been documented.22–24 The disease may also present in pregnancy and in this setting it is associated with an underlying disease process in about 50% of the cases.25–28 Large, necrotic ulcers, often 10 cm or more in diameter, characterize the disease (Fig. 15.1). Lesions may arise from acneiform pustules or on a background of erythematous nodules. Typically, the ulcers have undermined edges and red–purple borders (Fig. 15.2). They may be solitary or multiple, and occur most often on the lower limbs, although other sites such as the trunk, face, arms, and buttocks are sometimes affected (Fig. 15.3).29–30 Rare sites of involvement include the oropharyngeal region, hand, eyelid, eye, vulva, penis, scrotum, and the cervix.31–44 The ulcers are painful and tender, and may persist for months or years. Complications usually result from the site of the lesion and include cranial osteolysis and nasal perforation.17,45 Recurrent attacks are not uncommon.2 Cribriform scarring often follows healing. Systemic involvement has rarely been documented, affecting the lungs, liver, bone, joints, pancreas, and heart.46–51
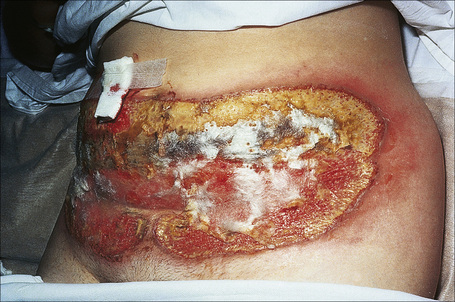
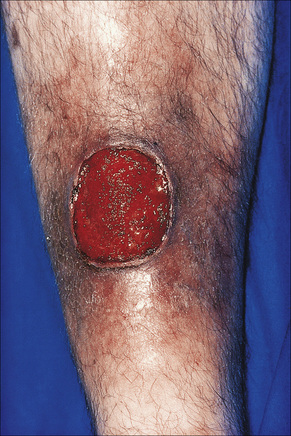
Fig. 15.2 Pyoderma gangrenosum: this shows an area of ulceration with a typical undermined purplish border.
By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
Occasionally bullous or pustular variants are encountered.4,52–60 One large study found that bullous lesions are more common on the upper extremities and they appear to be more frequently associated with hematological malignancy.4 Such lesions are sometimes designated atypical pyoderma gangrenosum.4,53 A vegetative form has also been described.61–65
A particularly interesting feature seen in as many as 50% of cases is development of lesions in traumatized areas (pathergy).4,66,67 Lesions may occur at sites of surgery and have been reported after cholecystectomy, breast reduction or augmentation, splenectomy, hysterectomy, cesarian section, following aortic valve replacement, at the site of a fasciocutaneous flap, a laparoscopic port insertion site, and in an amputation stump (Fig. 15.4).68–81 They also occur following rather trivial trauma such as injection or intravenous access site, arteriovenous dialysis shunt, blood-drawing, acupuncture or a tattoo.4,82–85 Pressure from use of seat belts in automobiles has been associated with subsequent lesion development.86 Presentation has even been documented at the location of a spider bite.87 One case reports involvement of the scalp after receiving hair highlights at a salon, which could be due to physical and/or chemical trauma.88
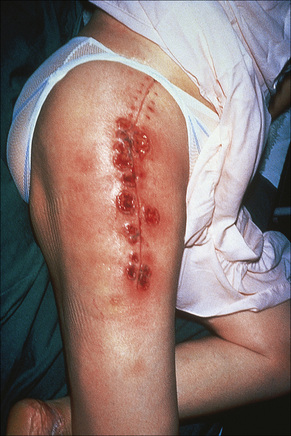
Fig. 15.4 Pyoderma gangrenosum: multiple early lesions at the site of previous surgery.
By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
A few publications have implicated drugs in the etiology of the disease, including:
In a single case report, pyoderma gangrenosum developed after combination therapy with cytosine arabinoside, aclarubicin, and granulocyte colony-stimulating factor for myelodysplastic syndrome.95 Another single report associates gefitinib, an EGFR inhibitor, with pyoderma gangrenosum.96 A granulomatous and suppurative dermatitis that may mimic pyoderma gangrenosum has been documented at the site of interferon-alpha (IFN-α) injections.97 Sclerotherapy has also been complicated by pyoderma gangrenosum on rare occasions.98 Association with sunitinib was reported in a single case study.99
Of particular importance is the known association of pyoderma gangrenosum with a variety of conditions4,11,100 (in up to 50% of patients2) as outlined in Table 15.1. Of these, inflammatory bowel disease (both Crohn’s disease and ulcerative colitis) and arthritis show the most well-established links.101–103 Pyoderma gangrenosum is reported to complicate around 1–2% of inflammatory bowel disease patients.104–106 In one study, 27% of patients had associated inflammatory bowel disease and 20% of patients had arthritis.4 In this same study, 27% of patients with superficial ‘atypical’ pyoderma gangrenosum had an associated hematological disorder.4 In another large study, idiopathic pyoderma gangrenosum and that associated with chronic inflammatory bowel disease were found to be more common in females, whereas pyoderma gangrenosum associated with hematological malignancy was more common in males.5 While pyoderma gangrenosum can fluctuate with inflammatory bowel disease activity, it may also be a presenting or heralding feature. The presence of pyoderma gangrenosum as a complication of inflammatory bowel disease does not appear to be an independent predictor of severity of the bowel disease.107,108 Although pyoderma gangrenosum lesions may improve as inflammatory bowel disease is brought under control, specific treatment is usually required.109 Most of the other numerous, rare associations mentioned in Table 15.1 are likely to be fortuitous, as a report of simple coexistence does not strictly imply a meaningful association.110 In any event, a diagnosis of pyoderma gangrenosum should always prompt an evaluation for an underlying disease association. Pyoderma gangrenosum-like lesions have been reported as the presenting feature of antiphospholipid antibody syndrome.111 Other underlying conditions causing lesions that mimic pyoderma gangrenosum are also well described including inflammatory and infectious processes, vasculopathies, and malignancies, stressing the importance of careful clinical investigation.112–122
Table 15.1 Conditions associated with pyoderma gangrenosum (alphabetical order)
The disease may also occur in association with other neutrophilic dermatoses including Sweet’s syndrome.123–125
Pyoderma gangrenosum is one of the components of a recently described autosomal dominant syndrome known as PAPA (pyogenic sterile arthritis, pyoderma gangrenosum and acne).126 This syndrome has been mapped to chromosome 15q and is associated with mutations in the gene CD2BP1/PSTPIP1.127–131 These mutations increase the binding affinity of this gene product to pyrin, overcoming the autoinhibition of this homotrimer and allowing activation of the downstream innate immune response.132–134 Ultimately, this mutation leads to an increase in caspase-1 activation, an underlying feature of multiple inherited autoinflammatory syndromes.135 Cases lacking a mutation in this gene have also been reported.136 These finding indicate that pyoderma gangrenosum may be best regarded as an autoinflammatory or autoimmune disease and the pathways being studied in PAPA syndrome may also eventually shed light onto the pathogenic mechanisms of sporadic pyoderma gangrenosum.
Para- and peristomal involvement in patients with ileostomy or colostomy for inflammatory bowel disease is a well-recognized phenomenon.4,137–141 In a large series, 13% of patients had peristomal pyoderma.4 Both Crohn’s disease and ulcerative colitis are associated with this complication.4,140,142 It should be noted that peristomal pyoderma gangrenosum has been seen in the absence of inflammatory bowel disease.137,138 It has been documented in patients with ostomy for gastrointestinal carcinoma and diverticular disease.137,138 Pyoderma gangrenosum may also occur at urostomy sites following cystectomy for bladder carcinoma.138
Superficial granulomatous pyoderma is believed to represent a superficial and rare variant of pyoderma gangrenosum.3,143–148 Patients develop single or sometimes multiple superficial ulcerated lesions with vegetative borders (for this reason this variant is sometimes referred to as ‘vegetative variant of pyoderma gangrenosum’) as a consequence of trauma, frequently surgical (Fig. 15.5). Pain is an occasional feature. The ulcers have a cleaner base than those seen in classic pyoderma. Lesions are most commonly found on the trunk and upper extremities and heal with cribriform scarring (Fig. 15.6). Draining sinuses are occasionally evident. Often there is no evidence of underlying systemic disease. Superficial granulomatous pyoderma is more likely to follow a chronic course compared with classic pyoderma gangrenosum.149
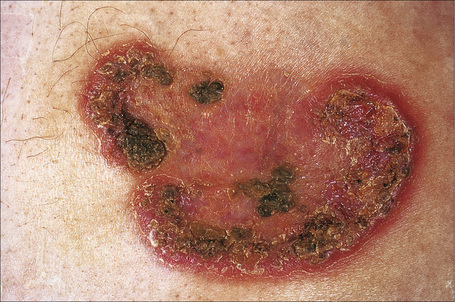
Fig. 15.5 Superficial granulomatous pyoderma: crusted superficial lesion with a cribriform appearance.
By courtesy of the Institute of Dermatology, London, UK.
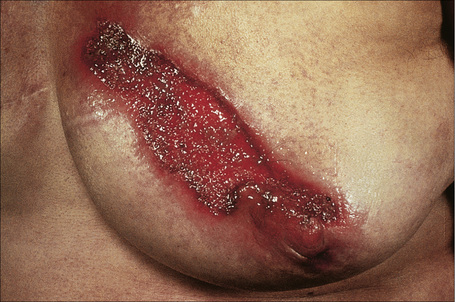
Fig. 15.6 Superficial granulomatous pyoderma: this field shows extensive ulceration of the breast.
By courtesy of R.K. Winkelmann, MD, Mayo Clinic, Scottsdale, Arizona, USA.
So-called ‘malignant pyoderma’ is a controversial designation which we believe should be avoided. Some authors have used the term to describe a variant of pyoderma gangrenosum predominantly affecting the head and neck.150–152 More recently, however, it has been postulated that at least some cases of so-called malignant pyoderma more likely represent cutaneous Wegener’s granulomatosis.153
One study found that over 50% of patients with pyoderma gangrenosum required long-term therapy to control their disease.5 The disease may be fatal in some cases, particularly if diagnosis is delayed.6,154 In another study, 2 of the 21 patients reported died of pyoderma gangrenosum secondary to pulmonary involvement.6
Pathogenesis and histological features
The precise pathogenesis of pyoderma gangrenosum is uncertain. The current state of knowledge suggests that it is due to immune dysfunction, perhaps innate, and/or that it develops on a vasculitic basis.10,128,155–160 A variety of immunological abnormalities have been described including:
The results of immunofluorescence studies in large series of patients have revealed both immunoglobulins (usually IgM) and complement in blood vessel walls in the dermis of the leading edge of the ulcer.175,176 Another study, however, failed to substantiate this finding.2 There is no evidence to support an infective pathogenesis.177
Expanding T-cell clones in both the skin and circulation have been described in a small series of patients indicating that T-cell response plays a role in the disease and may be triggered by a local stimulus in the skin.178 T-cell clonality has been described in pyoderma gangrenosum in the absence of an underlying myeloproliferative disease.179
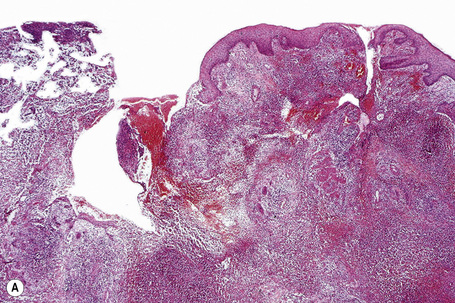
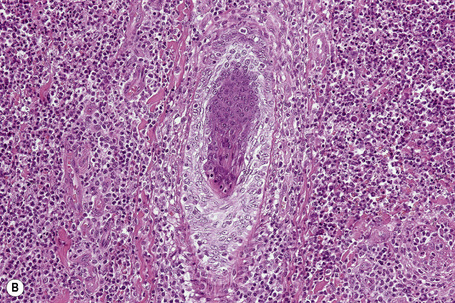
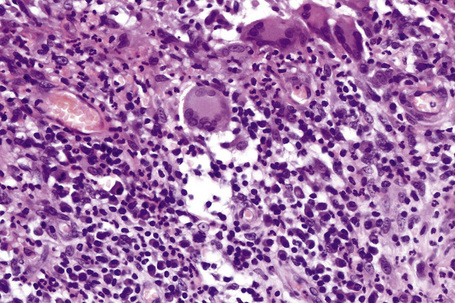
In general, the histopathology is that of non-specific ulceration with abscess formation (Fig. 15.7). The adjacent dermis shows acute and chronic inflammation. A pseudo-Pelger-Huet phenomenon with hyposegmented neutrophils making recognition difficult has been described in one patient.180 Early lesions may present with subcorneal pustulation (Fig. 15.8). Although the histological features of both leukocytoclastic and lymphocyte-mediated vasculitis have been described, it is our experience that any vasculitis present is usually located within the floor of the ulcer or in the immediate adjacent tissues and is, therefore, more likely to be a consequence, rather than a cause, of the lesion (Fig. 15.9).1,181
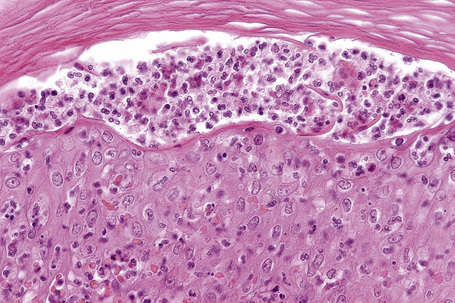
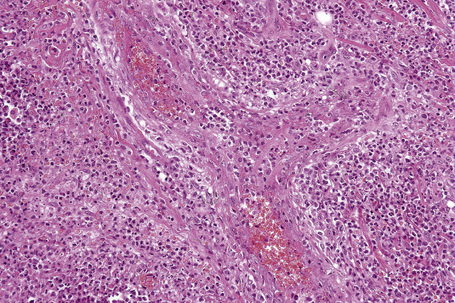
Giant cells appear to be a common feature of pyoderma gangrenosum in patients with Crohn’s disease.7 In one study, they were present in 6 of 13 patients with associated inflammatory bowel disease; of these, 5 had Crohn’s disease and 1 had ulcerative colitis.7 Giant cells were not found in any biopsies from 22 patients without associated inflammatory bowel disease.
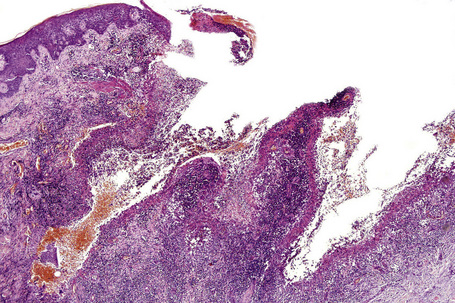
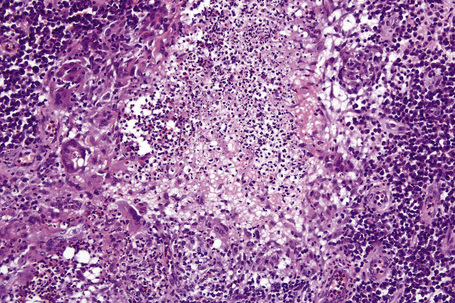
Superficial granulomatous pyoderma is characterized by a zoned inflammatory infiltrate in the superficial dermis.3 Focal and sterile abscesses are surrounded by a zone of granulomatous inflammation bordered by a rim of lymphocytes and plasma cells (Figs 15.10–15.12).3 Hemorrhage is often present and eosinophils may be evident. Any vasculitic change is thought to be secondary. The adjacent tissues may show scarring. Acanthosis and pseudoepitheliomatous hyperplasia are frequently noted. Foreign material including starch, sutures, vegetable matter, wood, and hair has been identified in a large proportion of these cases.3 It should be noted that not all cases of pyoderma gangrenosum with granulomatous inflammation are limited to the superficial dermis. Some cases show involvement of the deep dermis and even subcutaneous tissue.
Differential diagnosis
The histopathological findings in pyoderma gangrenosum are non-specific and the diagnosis is primarily one of exclusion.182 Since surgery is used to manage some of the disorders considered in the histological and clinical differential diagnosis – but is contraindicated in the treatment of pyoderma gangrenosum – early and accurate diagnosis is critical. Surgery, which tends to exacerbate the disease, is generally contraindicated in pyoderma cases because of the pathergic response. The mainstay of therapy is medical management, such as corticosteroids. Unfortunately, patients with pyoderma are often misdiagnosed early in the course of their disease and the diagnosis is sometimes made only after multiple unsuccessful (and damaging) surgeries have been performed. In one study, an average of five physicians had examined the patient before a correct diagnosis was rendered.29 To avoid this error, obtaining accurate clinical information on wounds and debridement specimens is essential.
Culture is required to exclude infection (bacterial, mycobacterial, fungal). Necrotizing fasciitis tends to affect deeper fascial and subcutaneous tissue, while pyoderma is centered in the dermis (albeit some spillover into the subcutis may be seen). Usually, sheets of bacteria are evident in untreated necrotizing fasciitis. Distinguishing these two conditions is critical since the treatments are diametric opposites with surgery and antibiotics for necrotizing fasciitis and avoidance of surgery with systemic anti-immune treatment and supportive wound care for pyoderma gangrenosum.183
Although some authors have noted lymphocytic or neutrophilic vasculitis in lesions of pyoderma gangrenosum, this finding, in our experience, is limited to areas adjacent to the ulcer and likely represents a secondary finding.5 Indeed, it has been our experience that ‘secondary’ vasculitis is frequently present at the border of ulcers of many different etiologies in patients without any genuine underlying ‘primary’ vasculitic process. Evaluation for vasculitis as a cause of ulceration therefore depends upon examination of blood vessels in areas of dermis and subcutaneous tissue away from the ulcer.
Acute febrile neutrophilic dermatosis
Clinical features
Acute febrile neutrophilic dermatosis (Sweet’s syndrome) is an uncommon disease of unknown etiology and pathogenesis.1–11 It is associated with a marked female predilection (5:1) and most patients affected are in their third through sixth decades. It may, however, occasionally be seen in children and a few cases presenting in infancy have been documented.12–22 Infant brothers who both had Sweet’s syndrome have been reported.23 Patients present with variable numbers of asymmetrically distributed, frequently bilateral, circumscribed, tender and painful red plaques or nodules, particularly on the face, neck, and upper and lower limbs (Figs 15.13–15.15). An acral form of this condition is now termed neurophilic (or pustular) dermatosis of the dorsal hands (Fig 15.16).24–31 Whether this represents a distinct disease or a peculiarly localized variant of Sweet’s syndrome is uncertain.
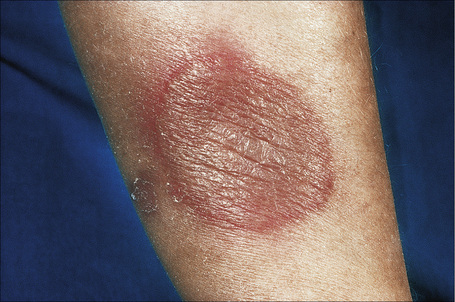
Fig. 15.13 Sweet’s syndrome: an erythematous plaque on the forearm.
By courtesy of R.A. Marsden, MD, St George’s Hospital, London, UK.
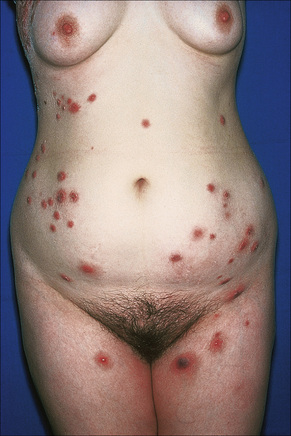
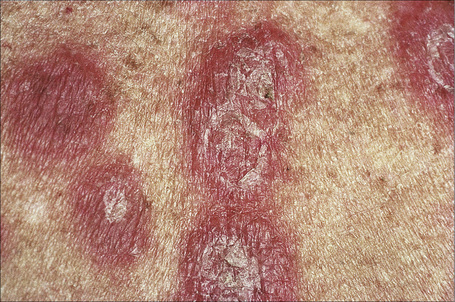
Fig. 15.15 Sweet’s syndrome: close-up view of typical plaques.
By courtesy of the Institute of Dermatology, London, UK.
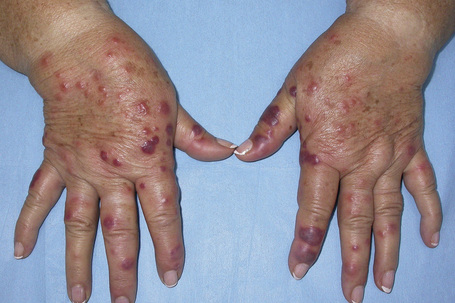
Occasionally, the lesions may become bullous or pustular.32,33 The plaques vary from about 1 to 4 cm in diameter and typically heal without scarring. Recurrences develop in approximately one-third of patients and postinflammatory hyperpigmentation is sometimes seen.34,35 Pathergy and koebnerization are occasional features and necrosis with ulceration may rarely be encountered.7,36–38 Sweet’s syndrome may present with lesions mimicking palmoplantar pustulosis and sometimes erythema nodosum-like lesions are present.32,39,40 A Sweet’s syndrome-like eruption has been described in association with exposure to light.41
Sweet’s syndrome often follows an upper respiratory tract infection. In some cases it is a complication of drug treatment, for example carbamazepine, furosemide, hydralazine, co-trimoxazole, abacavir, azathioprine, ofloxacin, doxycycline, clindamycin, minocycline, trimethoprim-sulfamethoxazole, bortezomib, lenalidomide, imatinib mesylate, nilotinib, etanercept, granulocyte colony-stimulating factor (G-CSF), radiocontrast agent, some vaccines, oral contraceptives, all-trans retinoic acid, isotretinoin, nitrofurantoin, diazepam, clozapine, and celecoxib.32,33,42–69 The temporal relationship with administration, development of symptoms, and resolution with withdrawal of the offending drug establishes the cause in drug-induced cases.42,65 The disease has also been reported after chemotherapy in patients with acute myeloid leukemia.70,71Sweet’s syndrome can be broadly reviewed as falling into three general categories:
Patients may also have conjunctivitis, episcleritis, iritis, polyneuropathy, oral involvement (superficial ulcers), and arthralgias.32,72–76 The larger joints are usually affected and involvement tends to be migratory.5 Patients with concurrent Sweet’s syndrome and erythema nodosum have been described and it is possible that these two disorders share common pathogenetic mechanisms.77 Dyssynchronous and synchronous Sweet’s syndrome and erythema nodosum may occur.78–81
Sweet’s syndrome is of particular importance since 10–40% of cases are associated with hematological malignancy such as leukemia (monocytic or myelomonocytic, including leukemia cutis), myelodysplasia, lymphoma, and multiple myeloma.82–93 Development of the disease may herald a relapse of the leukemia.94 Sweet’s syndrome has also been reported in patients with monoclonal gammopathy and myelodysplasia in the absence of frank leukemia or lymphoma.95 Hemophagocytic syndrome is also a reported association.96 The clinical lesions of Sweet’s syndrome are said to be more severe in patients with underlying hematological disease.88 An association with urticaria pigmentosa has also been documented.7
Solid tumors may also be associated with Sweet’s syndrome in up to 7% of patients, including:
Association with oral squamous cell carcinoma has been reported,36,100 as has a rare case following treatment of herpes simplex in a patient with metastatic breast carcinoma.101 Sweet’s syndrome has been described in conjunction with numerous conditions, some of which are listed in Table 15.2. While its association with hematologic and internal malignancies, upper respiratory tract infections, drugs, and certain inflammatory disorders such as erythema nodosum, rheumatoid arthritis, and sarcoidosis appears repeatedly, many of the others listed in the literature could be coincidental.
Table 15.2 Conditions associated with Sweet’s syndrome
| Common associations |
|---|
| Drugs58 |
| Hematologic malignancies (and myelodysplastic syndrome)91 |
| Hepatitis B142 |
| Inflammatory bowel disease (including ulcerative colitis and Crohn’s disease) 143,144 |
| Non-tuberculous mycobacterial infection145,146 |
| Pregancy147 |
| Sarcoidosis148,149 |
| Scrofuloderma150 |
| Sjögren’s syndrome151 |
| Tuberculosis152 |
| Upper respiratory tract infection10 |
| Rare associations |
| Bacille-Calmette–Guérin (BCG) vaccination153 Pigmented villonodular synovitis154 |
| Behçet’s disease155,156 |
| Bronchiolitis obliterans157 |
| Celiac disease158 |
| Chronic granulomatous disease159 |
| Dermatomyositis160,161 |
| Encephalitis162 |
| Erythema nodosum80 |
| Generalized granuloma annulare163 |
| Hashimoto thyroiditis164 |
| Infection with Apnocytophaga canimorsus, Chlamydia pneumoniae, Coccidiodes immitis, Cytomegalovirus, Francisella tularensis, Helicobacter pylori, Hepatitis C, human immunodeficiency virus (HIV), Pasteurella multocida, Salmonella enteritidis, and Staphylococcus epidermidis and Staphylococcus aureus165–178 |
| Polycythemia rubra vera179,180 |
| Prothrombin gene (G20210A) mutation181 |
| Relapsing polychondritis182,183 |
| Sarcoidosis184 |
| Solid tumor malignancy87,97 |
| Still’s disease185 |
| Surgery186 |
| Subacute and systemic cutaneous lupus erythematosus187–189 |
| Thyroid disease (Grave’s disease and Hashimoto’s thyroiditis)190 |
| Urticaria pigmentosa7 |
Systemic involvement may be a feature of Sweet’s syndrome with lesions described in the eye, lung, kidney, central nervous system, vagina, liver, gastrointestinal tract and skeletal muscle.7,86,102–110 Neural involvement appears to be strongly associated with HLA-Cw1.110 An exceptional case with gingival hyperplasia and myositis in the absence of cutaneous involvement has been documented.111
Associated features include pyrexia, neutrophilia, and a raised ESR. In one study, six of seven patients had antineutrophil cytoplasmic antibodies (ANCA).112 Other studies, however, have not found an association between Sweet’s syndrome and ANCA.7 This finding has not been further explored in the more recent literature.
Pathogenesis and histological features
The etiology of Sweet’s syndrome is unknown; however, the disease most probably represents an unusual hypersensitivity reaction.113 Cytokine mediation is likely, as anti-inflammatory agents are generally useful.113 The occasional presence of immune complexes in blood vessel walls may have pathogenetic significance.32 It has been suggested that neutrophils are activated by interleukin (IL)-1 and that Sweet’s syndrome represents a cytokine-mediated inflammatory reaction to a wide variety of different antigens including bacteria, viruses, drugs, and malignancies.114–116 Demonstration of elevated serum IL-1α, IL-1β, IL-2, and interferon-gamma (IFN-γ) but not IL-4 suggests that type 1 (but not type 2) helper T cells (Th) play a role in the pathogenesis.117 Not surprisingly, since exogenous treatment with G-CSF is associated with Sweet’s syndrome, endogenous G-CSF has been shown to be elevated in some cases.118
One study demonstrated clonality in the skin infiltrate of a patient with Sweet’s syndrome and acute myelogenous leukemia, undergoing treatment with G-CSF.119 The authors concluded that Sweet’s syndrome in patients with myelogenous leukemia may result from therapy-induced differentiation of neoplastic cells.119 However, a further study has demonstrated clonality in four patients in the absence of myeloproliferative disease.120
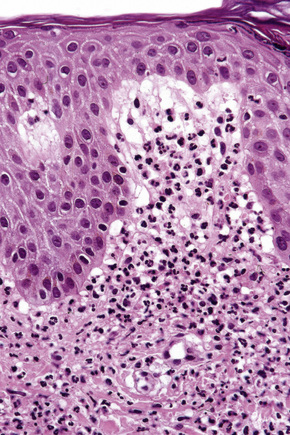
Histologically, the epidermis in Sweet’s syndrome is usually unaffected although occasionally slight spongiosis is present; rarely, vesiculation and spongiform pustules have been described.32 Necrotic keratinocytes are also sometimes evident.32 The cardinal feature, however, is an intense neutrophil polymorph infiltrate within the reticular dermis (Fig. 15.17).3,121 This may be diffuse or perivascular in distribution and often surrounds the sweat ducts. Typically, leukocytoclasis is marked (Fig. 15.18). Admixed with the neutrophil polymorphs are variable numbers of eosinophils, lymphocytes, and histiocytes. Ingestion of nuclear debris by histiocytes is sometimes a conspicuous feature. A histiocyte-rich form of the disease has increasingly been recognized.32,122–130 This is more likely to represent a stage in the evolution of the disease rather than a specific variant of Sweet’s syndrome. It has been postulated that the histiocyte-like cells seen represent immature granulocytes.130 Awareness of this form of presentation is important to avoid a misdiagnosis. Given the presence of a mononuclear cell infiltrate in these cases, it is extremely important to exclude leukemia cutis.
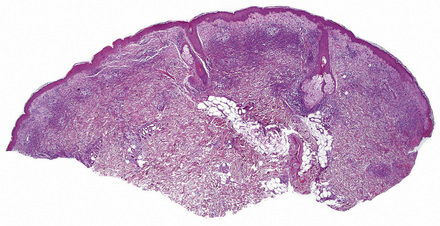
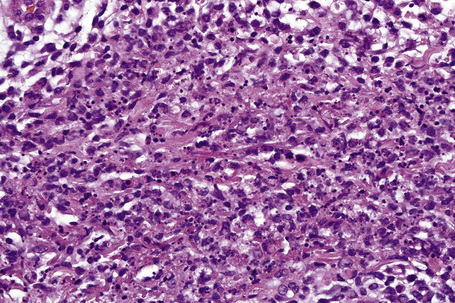
Often, the papillary dermis shows very marked edema, which sometimes results in subepidermal vesiculation (Fig. 15.19). Rarely, the presence of dermal papillary neutrophil microabscesses may cause diagnostic confusion with dermatitis herpetiformis (Fig. 15.20).121 In Sweet’s syndrome the blood vessels are dilated and may show endothelial swelling but changes of frank vasculitis are generally absent in our experience (and certainly not prominent). However, others have reported features of vasculitis such as nuclear dust, extravasation of erythrocytes, and fibrin deposition in and around vessels walls in the majority of their series of 31 cases and thus argue that the presence of vasculitis should not exclude this diagnosis.131–133 Purpura, however, is sometimes evident.134 Immunofluorescence examination of skin biopsies in Sweet’s syndrome is usually negative for immunoreactants in the walls of the vasculature. Recently, a case associated with leukocytoclastic neutrophilic lobular panniculitis has been reported.135
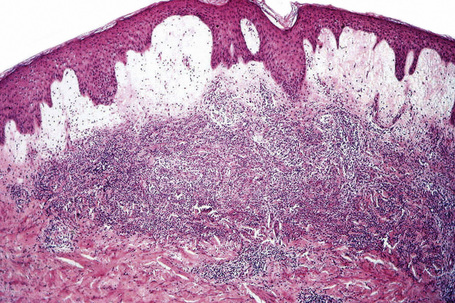
In some cases, an inflammatory infiltrate is noted within subcutaneous tissue. This infiltrate may be composed of lymphocytes and histiocytes and, less commonly, neutrophils.121,129 Sweet’s syndrome has also been associated with an erythema nodosum-like panniculitis.136 Elastophagocytosis and cutis laxa have been reported with resolution of Sweet’s syndrome lesions.137–140
Differential diagnosis
The presence of prominent fibrinoid vascular change can distinguish necrotizing vasculidities such as leukocytoclastic vasculitis, erythema elevatum diutinum, and granuloma faciale from Sweet’s syndrome; the clinical presentation and distribution of disease are also extremely helpful. In granuloma faciale, fibrinoid necrosis is often minimal and eosinophils tend to be prominent. Late lesions of erythema elevatum diutinum and granuloma faciale show fibrosis, a feature not seen in Sweet’s syndrome. Clinically, the presence of characteristic large ulcers helps distinguish pyoderma gangrenosum from Sweet’s syndrome. Also, pyoderma gangrenosum does not usually show the extent of karyorrhexis that is a typical feature of Sweet’s syndrome. A Gram stain and periodic acid-Schiff (PAS) or culture may be necessary to exclude infection. Distinction from some other forms of neutrophilic dermatosis including bowel bypass syndrome may be a definitional issue since the clinical setting determines the terminology applied.7 Behçet’s disease may also be associated with lesions similar to those seen in Sweet’s syndrome. Clinical correlation should ensure the correct diagnosis. CD30-positive forms can sometimes be found in Sweet’s syndrome, raising the possibility of lymphomatoid papulosis. However, neutrophils are usually rare in the latter condition and the number of CD30-positive cells in lymphomatoid papulosis is not prominent.141
Neutrophilic dermatoses associated with gastrointestinal and hepatobiliary disease
Clinical features
A syndrome of arthritis and pustular skin lesions was initially described in patients with inflammatory bowel and liver disease, and also in patients who have undergone jejunoileal bypass or Billroth II surgery for morbid obesity, but now has been described in other gastrointestinal disorders.1–7 Up to 20% of patients with jejunoileal bypass develop this condition.4,8 It has also been noted in association with peptic ulceration, appendicitis, and diverticular disease.9,10 An increasing number of papers call this process bowel-associated dermatosis-arthritis syndrome (BADAS); this terminology currently appears to be that preferred by the majority of authors.
The skin lesions may be papular or vesicular, or form large necrotic lesions resembling pyoderma gangrenosum. They are usually found on the trunk or extremities. Oral involvement has also been described.2 Associated panniculitis, which may resemble erythema nodosum, is a feature in some patients. Cutaneous manifestations often recur with exacerbation of the associated gastrointestinal disease.4 Some patients have an elevated ESR.4 The disease occasionally responds to antibiotic or steroid therapy.
A recurring vesiculopustular eruption may be seen in patients with hepatobiliary disease.11 The lesions – which can be pruritic and sometimes heal with an atrophic scar – often present on the extremities. In some patients the eruption represents a necrotizing folliculitis.11 Occasionally, the cutaneous lesions precede the features of the hepatobiliary disease.
Of interest, patients with Crohn’s disease complicated by disseminated abscesses (involving the spleen, lymph nodes, liver, pancreas, and brain) have recently been described.12 In some of these, the abscesses occurred before the diagnosis of Crohn’s disease. Histologically, a granulomatous element was commonly present. Successful treatment with immunosuppressive therapy suggests that such lesions may represent an unusual extraintestinal manifestation of Crohn’s disease.12
Pathogenesis and histological features
The presence of circulating immune complexes in occasional patients has led some authors to postulate a pathogenic role.13,14 It is postulated that bacterial overgrowth may play a role in the development of such circulating immune complexes.4
The histopathological findings, which are non-specific, are those of a neutrophilic dermatosis. The lesions show variable dermal edema and necrosis associated with a perivascular and interstitial neutrophilic infiltrate. Variable numbers of lymphocytes and histiocytes may also be present. Abundant karyorrhexis gives rise to a histological pattern similar to acute febrile neutrophilic dermatosis (Sweet’s syndrome) (Fig. 15.21). Leukocytoclastic vasculitis and pustular vasculitis have also been documented.15,16 The inflammation is often limited to the dermis but in some patients it may be seen to involve the subcutaneous fat, resulting in erythema nodosum or an erythema nodosum-like panniculitis.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


