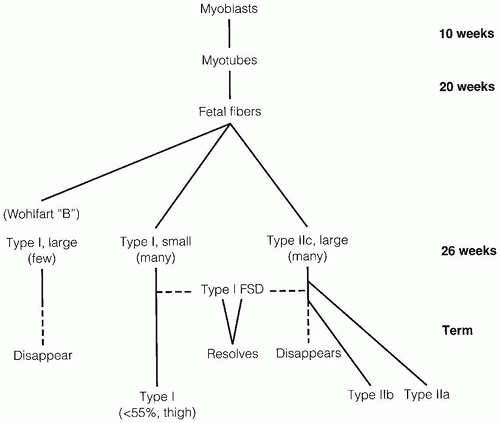
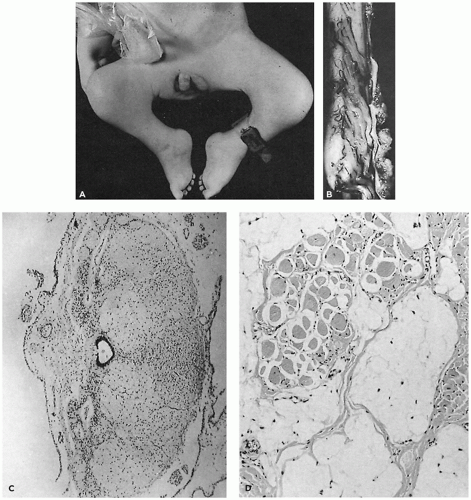
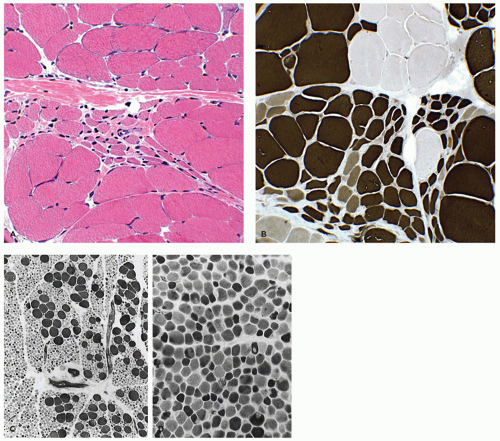
Detailed reviews of skeletal muscle development provide a basis for understanding early events in myogenesis (1,2). In the first few weeks of embryonic life (Figure 27-1), primitive mesenchyme, under the influence of MyoD, Myf, and PAX genes, differentiates to muscle progenitor cells. Myofibrils form under the influence of myogenin. Myoblasts with disorganized sarcomeres fuse to form multinucleate myotubes in which oriented sarcomeres surround a central core that is rich in organelles but devoid of contractile filaments (Figure 27-2A). At this stage, muscle nuclei are centrally located, and there are no definable histochemical or structural subtypes. Prominent peripheral nuclei are those of satellite cells, located within the sarcolemmal basement membrane, or residual unfused myoblasts. Desmin and myogenin are strongly expressed in fetal myotubes: this immunohistochemical reactivity is markedly reduced by late gestation (3). Further growth and development of muscle are influenced by workload, growth factors such as myostatin, and steroid hormones.
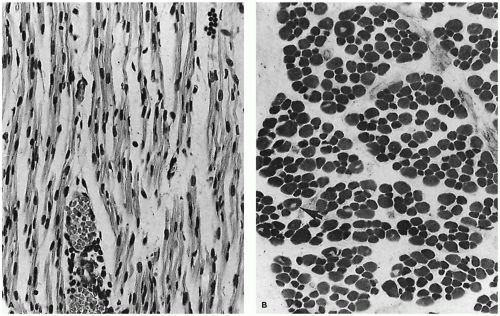
The transition from myotubes, which average 8 to 10 µm in diameter, to larger muscle fibers with peripheral nuclei is completed between 22 and 26 weeks of gestation when most are typeable as IIc fetal fibers containing fetal myosin. During the third trimester (Figure 27-2B), type IIc fibers are replaced by type I fibers, type IIa fibers, and type IIb fibers (4). Type IIc fibers, defined as those with an intermediate level of myosin ATPase reactivity at low pH, evolve to mature subtypes and finally disappear during early infancy. Abnormal persistence of type IIc fibers occurs in both myopathic and neuropathic disorders of infancy and may indicate a maturation disturbance. Myofibers containing fetal myosin may transiently reappear during regeneration after muscle fiber injury at all ages.
A few widely scattered, relatively large type I fibers (the Wohlfart B fibers) with an uncertain role in development make a brief appearance during late gestation and then rapidly regress. Most type I fibers are smaller than type IIc fibers until birth or slightly later in normal infants (5). Underlying the progressive disappearance of fetal fibers and appearance of mature fiber subtypes are poorly understood determinants of structural, enzymatic, and contractile proteins and of organelle populations including links between muscle and central nervous system maturation.
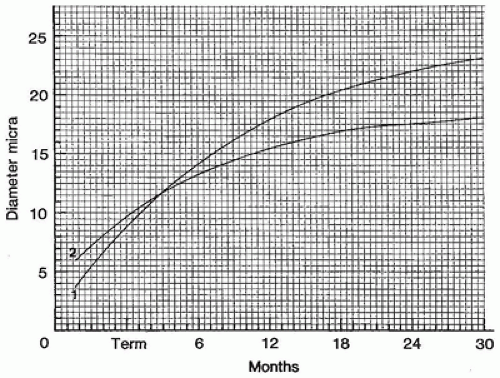
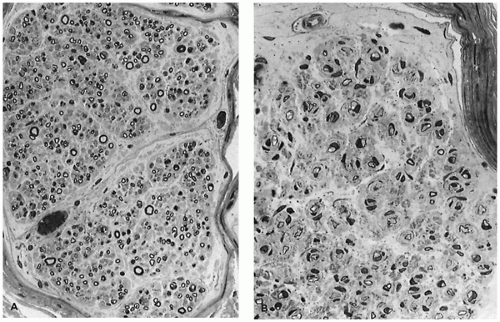
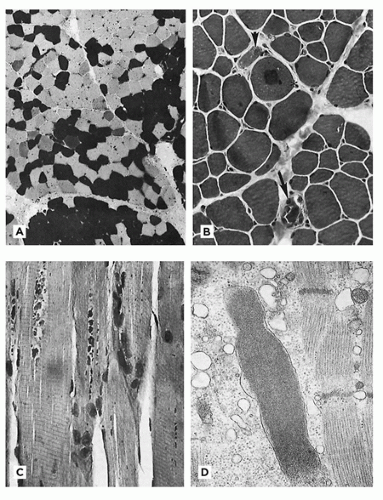
Brooke and Engel published a useful review of muscle fiber subtype profiles for normal and abnormal muscle in children (6). They identified disproportionately small type I muscle fibers as a major finding in childhood neuromuscular disease, defining this state as a difference in diameter of type I and type II fibers of greater than 12%. Their nomogram relating normal muscle fiber diameter to age is valuable but falls short of the ideal because data for several muscles and for fiber subtypes were pooled and because of the paucity of data from young infants. A nomogram derived from frozen postmortem specimens of thigh muscles (Figure 27-3) is useful for assessment of fiber size in infants (5). Muscle fiber diameter in formalin-fixed paraffin sections is about 70% to 80% of that in sections of fresh frozen muscle.
MUSCLE APLASIA AND HYPOPLASIA
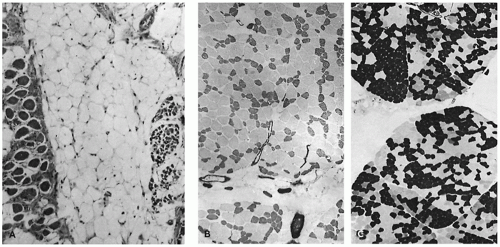
Determination of whether muscles are small because of primary failure to develop or because of regression is always problematic for the pathologist. The best documented examples of primary muscle aplasia or hypoplasia are secondary to spinal cord abnormalities. In the Poland anomaly, familial absent pectoralis muscle is associated with local soft tissue and skeletal defects and syndactyly (7). Möbius syndrome may coexist with the Poland anomaly, suggesting that a primary lesion may exist in the central nervous system and interfere with development of specific muscles in some cases (8). Chromosome abnormalities influence muscle development in trisomy syndromes, but it is uncertain whether this is a defect in muscle specification from primitive mesenchyme or the result of abnormal organization at the level of the spinal cord (9,10,11). The alleged normality of the spinal cord in most patients with urinary tract dilatation and deficient abdominal wall musculature (so-called prune belly syndrome) requires reexamination but focuses attention on alternate hypotheses for deficient numbers of fibers (Figure 27-4A), such as a mesodermal field defect, favored by the character of associated anomalies, and linkage to trisomy 18 (12). Compression secondary to abdominal distension may contribute to muscle atrophy by direct pressure, by limiting blood supply, or by interfering with innervation (Figure 27-4B, C).
FIGURE 27-1 • Steps involved in prenatal and perinatal muscle fiber maturation. Fsd, fiber size disproportion.
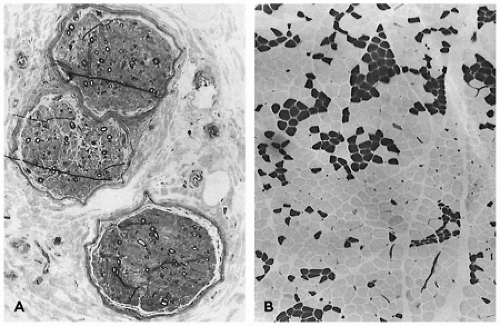
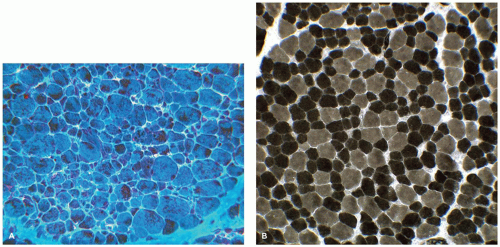
FIGURE 27-2 • A: Myotubular configuration of muscle fibers in a 19-week-old fetus. (Hematoxylin and eosin stain; original magnification ×400.) B: Normal deltoid muscle in a 29-week-old fetus contains small, dark, type I fibers and larger, intermediate-stained, type IIc fibers, many retaining central nuclei (arrow). (Myosin ATPase stain with preincubation at pH 4.3; original magnification ×250.)
FIGURE 27-3 • Diameter of normal type I (1) and type II (2) fibers at various ages in young children.
FIGURE 27-4 • A: Severely hypoplastic abdominal wall muscle in an infant with prune belly syndrome typically contains few fibers and abundant fat. (Hematoxylin and eosin stain; original magnification ×70.) B: Thin rectus abdominis muscle in a 19-year-old patient with prune belly syndrome exhibits type I FSD. (Myosin ATPase stain with preincubation at pH 4.3; original magnification ×80.) C: Thin rectus abdominis muscle in a 13-year-old child with prune belly syndrome. Homogeneous-type groups suggest reinnervation. (Myosin ATPase stain with preincubation at pH 4.6; original magnification ×400.)
Congenital diaphragmatic hernia is either an open defect or one covered only by a thin membrane lacking muscle fibers. Typically, located on the left, the phrenic nerve and the cervical spinal cord are normal. This form of hernia is thought to result from a primary defect in mesenchymal precursors involved in closure of the pleuroperitoneal canal. Eventration of the diaphragm is a unilateral or bilateral thin diaphragm that is a consequence of neuromuscular disease, such as birth trauma to the spinal cord or brachial plexus, anterior horn cell disease, phrenic nerve agenesis, myotonic dystrophy, or congenital myopathy (13). Diaphragm muscle usually displays the lesion of the primary disorder (see Chapter 12) (14,15,16).
ARTHROGRYPOSIS
Congenital joint contractures involving more than one area of the body (Figure 27-5A) usually result from intrauterine disorders that restrict fetal movement, also known as fetal akinesia deformation sequence (17,18,19). The etiology is heterogeneous and includes oligohydramnios, chromosome abnormalities of various types (20), teratogens, neurogenic and myogenic disorders, and rare restrictive skin and skeletal diseases (see Chapters 25, 26, and 28) (21,22). Most cases are sporadic and of unknown etiology. A tentative pathogenic classification (Table 27-1) of arthrogryposis congenita includes subtypes due to external compression, primary muscle disease, primary or secondary disorders of the central nervous system, such as dysgenesis or disruption of the motor centers of the brain or spinal cord, or deficiencies in the number or organization of the lower motor neurons and their peripheral connections.
FIGURE 27-5 • A: Congenital joint contractures due to distal spinal cord hypoplasia in caudal dysgenesis syndrome. B: The spinal cord in caudal dysgenesis is short and truncated, although it is histologically normal. C: Spinal cord in fetus with arthrogryposis multiplex is small, disorganized, and deficient in motor neurons. (Hematoxylin and eosin stain; original magnification ×64.) D: Thigh muscle in caudal dysgenesis is composed mainly of fat cells. A rare cluster of muscle fibers exhibits mixed hypertrophy and atrophy. (Hematoxylin and eosin stain; original magnification ×160.)
Specific neuromuscular diseases that cause contractures before birth usually are not associated with other major developmental defects. Notable exceptions are brain anomalies in patients with congenital muscular dystrophy (23), the Pena-Shokeir 1 phenotype, a lethal form of arthrogryposis congenita associated with pulmonary hypoplasia and diverse lesions in the nervous system (24), and multiple anomalies in some patients with a type II glutaric acidemia (25). The consequences of poor muscle tone or diminished movement vary from minor to life-threatening. Diminished fetal movement is one of several causes of short umbilical cord (26):
Short cord
Contractures
TABLE 27-1 PATHOGENETIC CLASSIFICATION OF ARTHROGRYPOSIS
Congenital arachnodactyly with contractures vs. Larsen syndrome
Restrictive dermopathy
External deformation (e.g., oligohydramnios)
Gracile ribs
Pectus excavatum
High arched palate
Laryngeal diaphragm
Hydramnios
Lung hypoplasia
Eventration
Banker proposed that arthrogryposis congenita multiplex is often the result of changes in muscle secondary to spinal cord disease (27). In a few arthrogrypotic fetuses, gross or microscopic malformation of the spinal cord is striking (Figure 27-5B, C), but the changes may be subtle (28). Autopsy studies after in-utero diagnosis indicate a higher incidence of brain than spinal cord abnormalities (19). The histology of poorly developed muscle related to congenitally fixed limbs is highly variable, which has engendered considerable confusion. Whether congenital arthrogryposis is due to lesions of the nervous system, muscle, or other tissues, muscle histology may be determined as much by the degree and duration of intrauterine immobility as it is by other factors (see Chapter 10). Careful selection of cases for muscle biopsy may increase the yield of diagnostic information (29). Changes are most likely in muscles contiguous with the contracture; when severe, fascial layers may enclose adipose tissue alone or with scattered muscle spindles or clusters of muscle fibers (Figure 27-5D). The hypoplastic muscle may collapse, producing condensation of stroma and an apparent increase in number of muscle spindles, often with extreme variation in muscle fiber size. Nonspecific myopathic changes seen focally may not be germane to pathogenesis. Recognizable denervation atrophy is uncommon. Theoretically, a marked deficit of muscle fibers could reflect primary deficiency of myoblasts, disturbed innervation at a critical early period, or loss of innervation due to circulatory disturbance or compression. Histologic criteria for establishing these distinctions are unreliable. It is probably best to avoid arbitrary designation of histologic changes as myopathic or neuropathic when the major abnormality is too few muscle fibers and too much adipose tissue. When in doubt, the term amyoplasia, used by clinicians to designate common sporadic form of ACM, may suffice.
Contractures may be a component of complex phenotypes of genetic origin, particularly when neurodevelopmental delay, developmental abnormalities, and/or facial abnormalities coexist (20). Pena-Shokeir phenotype (30,31,32,33) consists of central nervous system lesions, facial anomalies, polyhydramnios, fetal growth retardation, pulmonary hypoplasia, and arthrogryposis. The brain lesions in some examples are acquired disruptions (34,35). The occasional concurrence of Pena-Shokeir phenotype in siblings confirms that genetic factors are sometimes involved. Muscle may be normal, atrophic, or denervated. Reduced numbers of anterior horn cells are reported (33). Exposure of fetal rats to curare produces a similar phenotype, suggesting that fetal akinesia can produce structural anomalies of the face, polyhydramnios due to decreased swallowing, pulmonary hypoplasia due to decreased fetal breathing movements, and multiple joint contractures (18). Hall analyzed the Pena-Shokeir phenotype and agreed (17).
Primary muscle diseases that produce congenital contractures include the congenital myopathies (CMs) and the congenital muscular dystrophies. In specific forms of CM, contractures usually are not severe, the muscles are not hypoplastic, and muscle fibrosis, if present, is usually minor. In contrast, congenital muscular dystrophy is a diverse group of diseases that may be associated with contractures at birth or progressive contractures later in life, and prominent connective tissue proliferation in muscle.
DIAGNOSTIC EVALUATION
Muscle Biopsy
The muscle biopsy is a deceptively simple procedure, the success of which requires the cooperation of a clinician who determines the need and selects the muscle to be biopsied, a surgeon who has learned the skills requisite for a nontraumatized specimen, a specially trained histotechnologist, and a morphologist who interprets the findings in conjunction with the responsible clinician. Skeletal muscle biopsy plays a central role in guiding the management of a child thought to have a neuromuscular disease. Muscle biopsy should not be performed until other, noninvasive diagnostic methods have been used to evaluate the patient. These include a complete history (including birth and family history), physical evaluation by a neurologist, electrodiagnostic tests of neuromuscular function, and measurement of serum levels of enzymes, such as creatine kinase and aldolase, which are released by damaged skeletal muscle fibers.
Recommendations for muscle biopsy technique and processing are available from several sources (36,37,38,39). General anesthesia is necessary in small infants, but the risk of a malignant hypothermic reaction in patients with certain forms of muscle disease must be kept in mind. Local anesthesia may be used in older children, but the agent should not be directly injected into the muscle to be sampled.
Modified surgical biopsy using suction applied with a large-bore, reusable needle inserted through a small skin incision has been advocated as an alternative to open surgical biopsy. Applicable to patients of all ages except small infants, this technique produces a specimen of adequate size and quality for diagnostic studies for those neuromuscular diseases in which sampling error or precise orientation of myofibers is not an issue. Major advantages are use at the bedside or in an outpatient setting, low cost, ease of scheduling, and a small scar, all of which focus the decision to perform or repeat a muscle biopsy on essential medical criteria.
Optimal specimens obtained by open surgical biopsy are strips of fresh skeletal muscle, 1 to 2 cm long and approximately 0.5 cm wide, which are excised by sharp dissection. This is ideal for snap freezing and for preparation of frozen sections for histochemical procedures. If only one specimen is obtained, as in some infants with poorly developed muscles, a small portion of it can be excised from an untraumatized area before freezing and placed in an appropriate fixative for resin embedding and ultrastructural study.
Muscle biopsy clamps are not recommended because clumsy application creates troublesome artifacts from twisting, tearing, and compression, and a portion of the specimen is crushed.
A sample of every muscle biopsy performed should be prepared for ultrastructural studies, in case of need. If the status of the patient suggests a need for biochemical or molecular studies, an additional sample of fresh muscle should be obtained for immediate analysis or storage after snap freezing in liquid nitrogen. The specimen should be wrapped tightly to prevent desiccation.
Highly objectionable intracellular ice crystals form in slowly frozen skeletal muscle, because contractile proteins, when stained, provide a stark contrast to the angular spaces remaining after ice crystals have melted. Therefore, freezing skeletal muscle for histology requires liquid nitrogen or a halogenated hydrocarbon as the primary coolant. Isopentane is a secondary coolant to facilitate heat transfer from the muscle specimen. We find that a thick layer of talc applied to the muscle specimen prior to immersion in liquid nitrogen also is a good heat transfer agent. In our experience, freeze artifact is unavoidable in overly manipulated specimens, regardless of the method employed.
MUSCLE HISTOCHEMISTRY
Cross section is the preferred orientation of muscle fibers for diagnostic evaluation. In a minority of cases, useful supplementary information can be obtained by examining muscle fibers in longitudinal sections. Most laboratories no longer prepare paraffin sections of muscle because of the necessity of frozen sections for the diagnostic methods described below.
Among the dozens of histochemical and immunohistochemical methods that may be applied to skeletal muscle, a few are essential while the others are best reserved for exploring specific questions raised by the clinical impression or based upon the initial microscopical findings, such as metabolic myopathy, inflammatory myopathy, or muscular dystrophy. The modified trichrome method provides an overview, highlights collagen, displays normal and pathologic mitochondrial distribution, and reveals the myofibrillar network. Krebs cycle substrates, such as succinate, may be used to display mitochondrial activity and distribution, based on formazan deposition at sites of activity. DPNH reductase highlights areas of sarcoplasm lacking contractile elements. Alkaline phosphatase and reactions for fetal myosin assist in identification of injured or regenerating myofibers. Acid phosphatase helps to identify primary and secondary lysosomes.
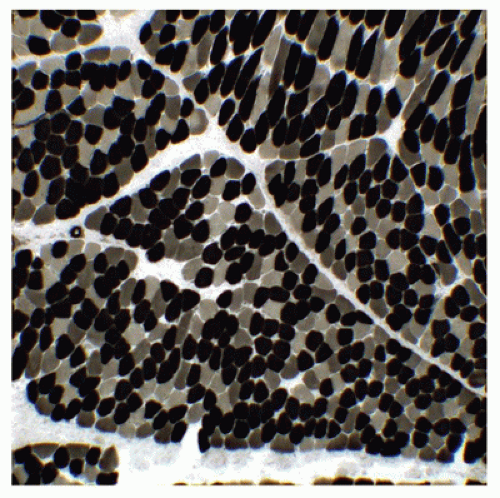
The calcium-dependent myosin ATPase reactions, performed by varying the preincubation substrate pH, are the mainstay for delineation of at least four major fiber subtypes in diagnostic muscle specimens (Figure 27-6). All human muscles are composed of a mosaic of fiber subtypes; the composition depends on the particular muscle or muscle group and its functions, the position within the muscle, constitutional differences between individuals, physical training, and disease. Although to some extent a dynamic quality, fiber subtypes revealed by the myosin ATPase reaction can be relied upon in most situations to provide information useful for diagnosis.
FIGURE 27-6 • Three major fiber subtypes are displayed by myosin ATPase reaction with preincubation at ph 4.6 in a normal thigh muscle from a 21-month-old girl. Type I fibers, dark; type IIa fibers, pale; type IIb fibers, intermediate density. (Persistent intermediate reaction after preincubation at pH 4.2 to 4.3 identifies type IIc fibers; original magnification ×200.)
Type I fibers are physiologic slow-twitch fibers that are richly endowed with mitochondria and contain relatively abundant, evenly dispersed lipid droplets, little glycogen, and lower concentrations of glycolytic enzymes, such as phosphorylase. Type II fibers are fast-twitch fibers that contain fewer mitochondria, relatively little lipid, abundant glycogen, and abundant phosphorylase activity. Fatigue-resistant muscles responsible for antigravity posture maintenance, such as the paraspinal and abdominal wall muscles, contain a high proportion of type I fibers. Muscles involved in rapid, repetitive movements, such as the intrinsic muscles of the larynx and the superficial muscles of the face, are predominantly composed of type II fibers. Among common biopsy sites, the deltoid and the gastrocnemius muscles have a predominance of type I fibers, and the quadriceps muscle has a predominance of type II fibers. Information on composition of less commonly biopsied muscles is available (40,41,42).
MUSCLE ATROPHY
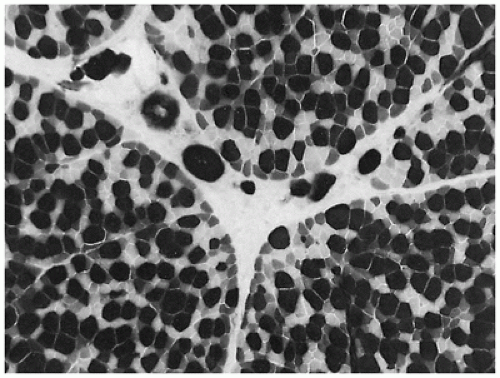
Loss of muscle bulk is a common feature of chronic neuromuscular disease in all age groups. In congenital muscle disorders, size of entire muscles and of individual muscle fibers may never have been normal. The term hypotrophy has been introduced to distinguish primary growth disturbance from secondary muscle fiber atrophy, but this distinction is difficult to make in practice. There is little information available on muscle fiber size in babies with intrauterine growth restriction. Muscle fiber atrophy tends to be uniform in malnourished infants (43). Muscle atrophy associated with central nervous system disorders, myasthenic syndromes, neoplasia, cardiomyopathy, inflammatory myopathy, or steroid therapy often selectively affects type II fibers (Figure 27-7). It has been suggested that type II fibers are more dependent than type I fibers on the trophic stimulation provided by normal activity levels and more vulnerable to the catabolic effects of steroids when activity is reduced (44).
FIGURE 27-7 • Selective atrophy of type IIa (pale) and type IIb (intermediate density) fibers in a child with idiopathic cardiomyopathy. (Myosin ATPase stain with preincubation at pH 4.6; original magnification ×400.)
DISTURBANCES OF MUSCLE MATURATION
Features of muscle immaturity include persistent myotubes, central nuclei, prominent satellite cells, type I fiber size disproportion (FSD), large isolated type I Wohlfart fibers, a high percentage of fetal type IIc fibers, numeric predominance of type I fibers, and absence of type IIb fibers (45). Signs of muscle maturation disturbance are common in infants with congenital myopathies, infantile spinomuscular atrophy (SMA), peripheral neuropathy manifested in infancy, and in central hypotonia (46). In the perinatal period, intramuscular myelopoiesis in perivascular connective tissue may accompany dysmaturation.
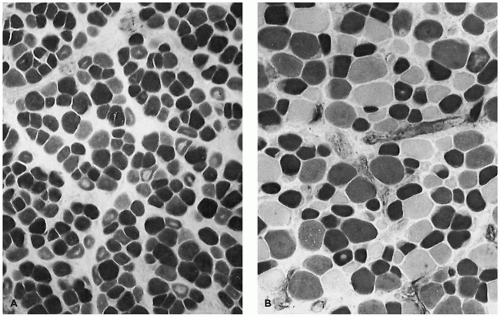
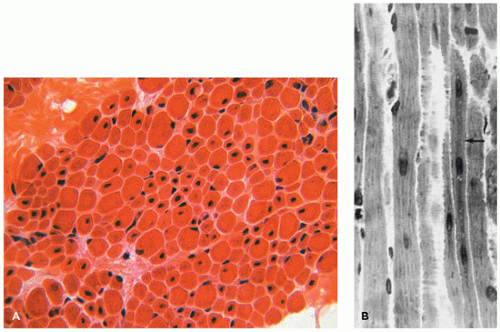
An important prototype for muscle maturation delay occurs in newborn infants of mothers with myotonic dystrophy, a dominantly inherited disease of variable severity. Even when severely affected, such infants usually improve clinically and in terms of muscle morphology (Figure 27-8). The causative mutation is an expansion of a trinucleotide repeat sequence in the myotonin gene, which is the basis for a reliable diagnostic test on blood DNA that has largely supplanted muscle biopsy. The degree of expansion correlates reasonably well with disease severity and tends to increase from one generation to the next, particularly in women.
CENTRAL HYPOTONIA
Central hypotonia designates a heterogeneous clinical group of infants and young children who display general poor muscle tone and various degrees of slowing of motor development accompanied by one or more signs of central nervous system dysfunction, such as motor delay, decreased alertness, delayed or dysarthric speech, seizures, learning disabilities, pyramidal tract signs, or ataxia. Some of these children have defined syndromes, but many do not. Central nervous system disorders associated with hypotonia and/or morphologic signs of muscle dysmaturity in infancy include
Microcephaly, macrocephaly
Ischemic encephalopathy
Cerebral palsy
Cerebellar hypoplasia
FIGURE 27-8 • A: Severe immaturity in thigh muscle of a term baby with myotonic dystrophy. Type I fibers and type IIc fibers prevail. Nonstaining central zones are myotubes or central nuclei. (Myosin ATPase stain with preincubation at pH 4.3; original magnification ×740.) B: Thigh muscle of an improved 3-month-old infant with myotonic dystrophy syndrome exhibits near-normal composition with persistent small type I fibers. (Myosin ATPase stain with preincubation pH at 4.3; original magnification ×800.)
Congenital muscular dystrophy
Cerebral dysgenesis
Imaging studies of the brain may be normal or show borderline abnormalities, including expansion of the subarachnoid space, atrophy of cortical gyri, polymicrogyria, ventricular dilatation, or cerebellar hypoplasia. Infants who are suspected of having central hypotonia are sometimes subjected to muscle biopsy for the purpose of ruling out specific causes of muscle hypotonia. Thigh muscle samples may be normal but often display signs of muscle dysmaturation with disturbances of fascicular composition, irregularities of fiber size, and distribution suggesting previous denervation and reinnervation or preferential atrophy or hypertrophy of type I or type II fibers (Figure 27-9) (47). A similar spectrum of changes has been described in specimens from spastic muscles in children with cerebral palsy (48). It is relevant that a history of perinatal asphyxia is obtained in some patients with central hypotonia, some of whom eventually develop spastic paresis (46).
Several authors have suggested that a suprasegmental determinant of normal skeletal muscle maturation may be disturbed in some infants with central hypotonia or arthrogryposis multiplex congenita (19). However, skeletal muscle development is usually quite normal in infants with anencephaly, indicating that normal unmodified spinal cord function is sufficient for lower motor unit maturation. It is possible that sporadic central hypotonia results in some babies from simultaneous perinatal insult to the developing central nervous system and the peripheral motor units. Alternatively, a genetic defect may affect both brain and muscle development, as in some children with congenital muscular dystrophy.
ROLE OF MUSCLE BIOPSY
Pathologic alterations in muscle fibers are conventionally classified as “myopathic” or “neuropathic” to signify, respectively, primary diseases of the muscle fibers and diseases of fibers as components of motor units. Many muscle specimens from infants or children show readily classifiable changes (Table 27-2), which correlate well with results of clinical investigations. However, troublesome discrepancies are common, requiring cooperation between the clinician and the morphologist, and considerable experience to resolve. The conventional myopathy versus neuropathy paradigm is often uncertain in congenital neuromuscular diseases. Detailed histochemical and ultrastructural investigation of muscle specimens from patients with neuromuscular dysfunction has increased dramatically the number of recognizable clinicopathologic entities. In recent years, the determination of the cellular and genetic basis for many neuromuscular diseases has in some instances replaced diagnostic histomorphology. However, muscle biopsy, appropriately performed and expertly evaluated, remains indispensable for precise diagnosis and for orientation of clinical thought in most cases (38).
FIGURE 27-9 • A: Thigh muscle in central hypotonia showing abnormal numeric predominance of type I fibers. (Myosin ATPase stain with preincubation at pH 4.6; original magnification ×160.) B: Thigh muscle in central hypotonia (postperinatal asphyxia) showing small type I fibers, singlets, and small clusters. (Myosin ATPase stain with preincubation at pH 4.6; original magni-fication ×160.) C: Thigh muscle in central hypotonia showing small type II fibers and hypertrophied type I fibers. (Myosin ATPase stain with preincubation at pH 4.6; original magnification ×400.)
TABLE 27-2 MUSCLE FIBER PATHOLOGY
Neuropathic Changes
Myopathic Changes
Changes of Indeterminate Cause
Motor unit distribution
Focal or general distribution
Muscle hypoplasia
No fiber subtype preference
May be influenced by fiber type
Delayed or arrested maturation
Poor fiber subtype display
Loose or compact grouped atrophy
Fiber degeneration, many forms
Central nuclei
Type I atrophy
Grouped fibers of similar subtype
Fiber necrosis, segmental
Type II atrophy
Angular atrophic fibers
Fiber regeneration
Extreme fiber atrophy with nuclear aggregates
Replacement fibrosis
Inflammatory reaction
Limited degeneration and regeneration
Organelle diseases
Acute atrophy
Storage diseases
Target fibers
Condensation of structural collagen
Table 27-3 outlines the primary neuromuscular diseases presenting in infancy and childhood. Six generic categories are delineated:
Spinal cord hypoplasia or dysplasia Spinomuscular atrophy
Type I (infantile, Werdnig-Hoffmann)
Type II (late infantile)
Type III (juvenile, Kugelberg-Welander)
Infantile spinomuscular atrophy, X-linked
Bulbar muscular atrophy (Fazio-Londe)
Scapuloperoneal spinal muscular atrophy
Hereditary sensory motor neuropathies, all types
Acquired myelopathy or neuropathy
Traumatic
Ischemic
Infectious (enterovirus)
Postinfectious (Guillian-Barré)
Toxic/drug-induced
Nutritional
DISORDERS OF INNERVATION
Denervation is expressed in all of the muscle fibers of an affected motor unit, producing clusters of hypotonic atrophic muscle fibers. Denervated fibers often exhibit angulated contours due to molding by normotonic neighbors. The appearance of denervated muscle has no specificity for the underlying neurologic disorder (Table 27-4) and may depend on the chronologic relation of the biopsy to the onset of the disease (Figure 27-10A, B). Findings are further modified by expression in utero or during early infancy, a time when impairment of muscle maturation may be added to the effect of denervation (Figure 27-10C, D). Reinnervation occurs unpredictably as the distal nerve twigs of an intact motor unit expand to make new junctions with adjacent denervated fibers, resulting in abnormally large groups of fibers of one subtype (Figure 27-9B). Recognition of clusters of reinnervated fibers in muscle that normally contains mostly one type of fiber may require a quantitative approach. Other helpful features of chronic denervation are target and targetoid fibers, and central nuclei (49).
Spinal Cord Diseases
Deficiency of spinal neurons results from developmental deficiency (dysplasia or hypoplasia) or may result from degeneration or loss caused by genetic motor neuron diseases, trauma, ischemia, and viral infections (e.g., enterovirus) with affinity for spinal motor neurons. Microscopic dysplasia or malformation of the spinal cord is typically sporadic and may be generalized or limited to the distal cord, causing segmental deficiency of lower motor units, muscle hypoplasia, and contractures (27).
Spinal cord trauma occurs during difficult deliveries or as a result of accidents, and it does not selectively damage the anterior horn regions. Diaphragmatic paralysis in newborns is more often the result of unilateral brachial plexus injury than of spinal cord injury. Cervical spine instability or a narrow cervical canal, as in skeletal dysplasia or other developmental disorders involving the spine, predisposes to hyperextension injury of the spinal cord. Acute selective upper spinal motor neuron necrosis occasionally accompanies widespread perinatal ischemic injury to brain stem nuclei (50). Spinal cord injury from compromise of the spinal arterial circulation is a recognized complication of surgical procedures near the aorta.
FIGURE 27-10 • A: Active denervation involves fibers of each subtype that causes atrophy, often with angulated profiles. (Hematoxylin and eosin stain; original magnification ×400.) B: Reinnervation produces clusters of fibers of similar type in chronic peripheral neuropathy. (Myosin ATPase stain with preincubation at pH 4.6; original magnification ×400.) C: Infantile spinomuscular atrophy (ISMA). Subtotal or panfascicular fiber atrophy with clusters of hypertrophied type I fibers. (Myosin ATPase stain with preincubation at pH 4.6; original magnification ×64.) D: Atrophic fibers in ISMA exhibit features of delayed maturation. (Myosin ATPase stain with preincubation at pH 4.3; original magnification ×640.)
MOTOR NEURON DISEASE
Hereditary motor neuron diseases of children are characterized by progressive degeneration of the motor neurons of the spinal cord. Typically, sensory and upper motor neurons are not involved. Classification is based on factors such as age at presentation, rate of progression, milestones achieved, and distribution of neuronal lesions (51). The following clinical classification enjoys wide usage:
Type 1: never able to sit, onset before 6 months (Werdnig-Hoffman disease)
Type 2: able to sit but not to walk, onset 6 to 18 months (Dubowitz disease)
Type 3: able to walk, onset in childhood beyond 18 months (Kugelberg-Welander disease)
Type 4: adult onset
Infantile SMA is characterized by rapid progression and may be recognized at birth, or the onset may be delayed. Weakness is generalized but mainly proximal; respiratory muscles are spared; diaphragmatic involvement is rarely observed at onset. Onset in utero (proposed Type 0) with fetal akinesia syndrome or arthrogryposis is very rare.
Infantile SMA has an incidence of 1/10000 and long has been considered an autosomal recessive trait. Linkage studies have now mapped the gene to a 5q deletion in greater than 95% of cases of type 1 SMA, including many families with a pattern of later onset (52,53). Important deletions have been identified in the survival motor neuron gene (SMN). Deletions in SMN are found in more than 90% of children with types 1 and 2 SMA and about 80% with type 3 SMA but are absent in patients with type 4 SMA. SMN1-negative variants of infantile SMA include a diaphragmatic form with early respiratory failure and distal muscle atrophy (SMARD1) due to mutation of a gene on 11p13 (54) and an X-linked arthrogrypotic form that also lacks the 5q/SMN deletion (55).
Genetic testing is a diagnostic tool in SMA, eclipsing the role of muscle biopsy, except when clinical findings are unusual or loss of function SMN1 mutations are negative. A cautionary note: the SMN gene deletion may be deleted in normal siblings. SMN deletion negative cases, though a small minority, when studied with next-generation sequencing technologies, have shown remarkable clinical and genetic diversity (53).
Muscle biopsy discloses groups or entire fascicles composed of small rounded fibers displaying poor delineation of fiber subtypes, small immature type I fibers, variable persistence of type IIc fibers, and small clusters of hypertrophied type I fibers (see Figure 27-10C, D). Features of infantile SMA in muscle may be confused with the incomplete muscle maturation that prevails in late gestation. SMA presenting in older children exhibits features similar to denervation in adults, including clusters of angulated atrophic fibers and groups of hypertrophied reinnervated type I and type II fibers. In older children, proximal muscle involvement in SMA may simulate muscular dystrophy. In addition to denervation atrophy, focal myopathic changes may be observed, as is true in other chronic denervating diseases. Single-fiber electromyography may help resolve the issue of pathogenesis in patients with mixed neuromyopathic biopsy findings by identifying enlarged motor units, a consequence of reinnervation.
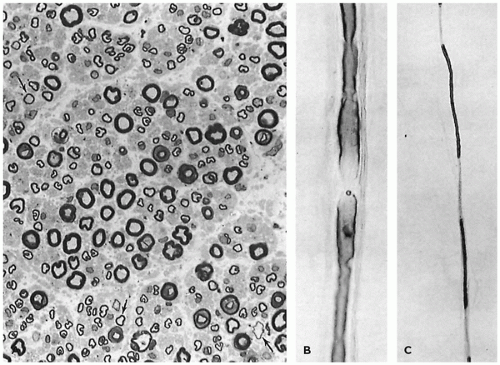
FIGURE 27-11 • A: Sural nerve in idiopathic childhood neuropathy, possibly type I hereditary sensory motor neuropathy (HSMN), with scattered “onion bulbs” and variable myelin thickness. (Methylene blue and azure II stain; original magnification ×400.) B: Sural nerve in type III HSMN with prominent “onion bulbs” and uniform severe defect in myelin thickness. (Methylene blue and azure II stain; original magnification ×640.)
PERIPHERAL NEUROPATHY
The diagnosis of the peripheral neuropathies in children is facilitated by electrophysiologic studies that distinguish myelinopathies from axonopathies, by evaluation of individual teased axons, by light and electron microscopy of sural nerve specimens, and by genetic testing for more than 70 genes that have been linked to Charcot-Marie-Tooth disease with weak phenotype-genotype correlation (56,57).
Hereditary sensory motor neuropathies (HSMN) are far more common in children than acquired chronic neuropathies. Classic phenotypes include the usually autosomal dominant hypertrophic peroneal neuropathies of Charcot-Marie-Tooth (types I and II hereditary sensory motor neuropathy), the autosomal recessive hypertrophic hypomyelinating infantile neuropathy of Dejerine-Sottas (type III hereditary sensory motor neuropathy), and Refsum disease (type IV hereditary sensory motor neuropathy).
Schwann cell proliferation that results in “onion bulb” formation (Figure 27-11A, B) is most prominent in type III, but it is not a specific feature for genetic disorders. Delayed nerve conduction velocity related to myelination disorder is a feature of all except type II. Denervation of muscle may occur with axonopathy. Genetic analyses have disclosed extreme heterogeneity with defects in different steps in myelin formation producing overlapping phenotypes. Neuropathy in infants with extreme hypomyelination and Schwann cell proliferation associates with multiple gene defects. Congenital absence of peripheral myelin is described in a lethal form of arthrogryposis congenita (58) and has also been observed in some infants with congenital muscular dystrophy (59).
FIGURE 27-12 • A: Sural nerve in prolonged Guillain-Barré syndrome. Thin myelin sheaths are inconspicuous. There is no inflammation. (Methylene blue and azure II stain; original magnification ×640.) B: Teased isolated myelinated fiber. Demyelination begins at nodes of Ranvier. C: Teased isolated myelinated fibers with segmental demyelination, typical of postinfectious polyneuropathy.
Acquired demyelinating diseases of peripheral nerves include acute postinfectious polyneuritis (Guillain-Barré syndrome) and chronic idiopathic inflammatory polyneuropathy (60,61). Both cause hypotonia, weakness, hyporeflexia, and slow nerve conduction, more commonly in older children or adults and rarely in infancy. In both conditions, segmental demyelination and remyelination are accompanied by Schwann cell proliferation and mononuclear infiltrates of variable severity (Figure 27-12). Inflammation is often scanty, making distinction from genetic forms of peripheral neuropathy difficult. Perivascular infiltrates of macrophages may be helpful (62). In chronic cases, muscle wasting probably is due more to disuse than to denervation, which typically is lacking. Peripheral neuropathy caused by drugs, heavy metal intoxication (e.g., lead), or bacterial toxins (e.g., diphtheria) may be due to axonal degeneration alone or combination with demyelination.
COMBINED CENTRAL AND PERIPHERAL NEUROPATHY
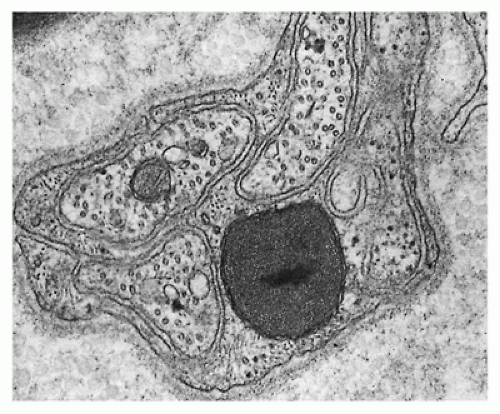
Complex multisystem metabolic diseases and genetically determined diseases of central neurons or central white matter usually do not involve peripheral nerves, but exceptions are noteworthy. Rare patients with mitochondrial myopathy and peripheral neuropathy have complex crystalline inclusions in Schwann cell cytoplasm (Figure 27-13) (63). Lower motor neuron involvement in olivospinocerebellar atrophy, Friedreich ataxia, and ataxia-telangiectasia leads to axonal loss and muscle denervation and reinnervation (Figure 27-14), which usually is overshadowed clinically by the peripheral and central sensory deficits. Neuroaxonal dystrophy involves central and peripheral axons in which focal expansile lesions accumulate complex tubulomembranous inclusions. Among the leukodystrophies, the metachromatic subtypes are most likely to involve peripheral nerves and may cause muscle denervation; peripheral neuropathy also occurs in Krabbe disease (see Chapters 5 and 10).
FIGURE 27-13 • A Schwann cell, enclosing several unmyelinated nerves, contains crystalline inclusion with double mitochondroid outer membrane. Patient had abnormal mitochondria in heart, skeletal muscle, and peripheral nerves. (Uranyl acetate and lead citrate stain; original magnification ×20,000.)
FIGURE 27-14 • A: Marked reduction of fascicular area and number of myelinated axons in the sural nerve from an older child with Friedreich ataxia. (Original magnification ×200.) B: Denervation atrophy with type groups indicating reinnervation in Friedreich ataxia. (Myosin ATPase stain with preincubation at pH 4.3; original magnification ×64.)
FIGURE 27-15 • A: Reinnervation (type groups) in a child with chronic cholestasis from infancy causing central and peripheral neuropathy due to low levels of vitamin E. (Myosin ATPase stain with preincubation at pH 4.6; original magnification ×160.) B: Mixed neuromyopathic change in a child with prolonged vitamin E deficiency. Fiber necrosis (arrow), degeneration (arrowhead), and target fibers (asterisk) are shown. (Trichrome stain; original magnification ×500.) C: Massive deposition of residual bodies in a child with chronic cholestasis and low vitamin E levels. (Methylene blue and azure II stain; original magnification ×1,000.) D: Dense monomorphous residual body, typical of infantile vitamin E deficiency myopathy. (Uranyl acetate and lead citrate stain; original magnification ×10,500.)
NUTRITIONAL DISORDERS
Neuropathies due to malnutrition are rare in industrialized societies. However, deficient absorption of fat-soluble vitamins may produce neuropathy in infants with chronic cholestasis, in cystic fibrosis, or in abetalipoproteinemia. Areflexia or progressive ataxia due to central dysfunction due to vitamin E deficiency complicates unrelenting cholestasis in early infancy (64). In these children, muscle fiber-type groups develop, residual bodies accumulate in muscle fibers (Figure 27-15) and in Schwann cells, and serum creatine kinase may be elevated. This mixed neuromyopathy can be arrested with parenteral vitamin E therapy (65).
DISORDERS OF NEUROMUSCULAR TRANSMISSION
The neuromuscular junction is the target of many toxic, infectious, or autoimmune insults and the locus of several rare genetically transmitted defects. The specific site of impairment may be presynaptic or postsynaptic. The differential diagnosis of these conditions requires evaluation of response to acetylcholine esterase inhibition, detailed electromyography, measurement of circulating antibodies to acetylcholine receptors, and in selected cases, morphologic studies of motor end plates, which are best sampled in external intercostal muscle specimens. Muscle may be normal or show selective type II fiber atrophy or overt signs of denervation.
Neonatal myasthenia gravis is a transient condition expressed within the first few days after birth and continuing for several weeks thereafter in babies born to mothers with myasthenia gravis. Affected infants are otherwise normal, although the rare coexistence of congenital contractures suggests the possibility of in utero injury. The mediator of the disease is thought to be transplacental maternal IgG antibody to acetylcholine receptors on the postsynaptic muscle membrane. High maternal levels of pathologic antibody increase the risk. Myasthenia gravis, similar to that in adults, also occurs in older children. Antibody to acetylcholine receptor is usually demonstrable, and acetylcholine esterase inhibitors relieve symptoms. Congenital myasthenia syndromes are rare heterogeneous genetic disorders of neuromuscular transmission that cause hypotonia, weakness, ptosis, respiratory problems, and motor delay (66). Diagnosis is challenging and often delayed. Muscle biopsy is usually not helpful.
Acquired disorders of neuromuscular transmission in children are caused by neurotoxins associated with diphtheria or the infantile form of botulism, drugs such as magnesium sulfate or aminoglycoside antibiotics, and neoplasms or autoimmune disorders that cause motor conduction changes typical of the Eaton-Lambert syndrome. Infantile botulism is of particular interest because it results from endogenous production of a neurotoxin by gastrointestinal flora rather than ingestion of preformed toxin. Early signs may be constipation and various degrees of failure to thrive. In protracted cases, dysphagia, loss of head control, and progressive flaccid paralysis develop. A few infants with sudden unexplained death have had clostridial endotoxin detected in the gut. The basis for transient susceptibility of infants to endogenous toxin production is unknown. Morphologic studies of terminal motor nerves in affected infants have not been reported.
Serious neuromuscular sequelae to intensive care may be due to excessive use of curare-like drugs, to concomitant exposure to aminoglycoside antibiotics or to corticosteroid drugs, or to severe underlying illness, such as sepsis. In all age groups, a few patients supported by prolonged exposure to curare-like drugs during mechanical ventilation develop persistent weakness or after withdrawal (67). The consequences may be profound, especially in infants (68,69). Autopsy evaluation of psoas muscle fiber diameter in infants who had been paralyzed for extended periods indicated smaller average muscle fiber diameter than expected, suggesting the possibility of muscle fiber growth retardation (68). Muscle biopsy may help exclude antecedent neuromuscular disease and identify the basis for prolonged weakness. Clinically overlapping disorders include critical illness polyneuropathy and critical illness myopathy.
CONGENITAL MYOPATHY
The congenital myopathies (CMs) are static or slowly progressive disorders of muscle of genetic origin that exhibit substantial clinical overlap. Definitions based on distinctive morphology may be supplemented by knowledge of mutations in proteins such as alpha-actin, alpha-actinin, nebulin, beta-tropomyosin, or myotubularin (70,71). Clinical severity correlates imperfectly with morphologic features and specific gene defects. Weakness, first detected at any age, is associated with, normal serum levels of creatine kinase, absence of electrophysiologic or morphologic evidence for denervation, and no other definable primary muscle diseases, such as a dystrophy or myositis. Subtypes with most distinctive morphologic features are myotubular-centronuclear myopathy, nemaline myopathy, central core disease, minicore-multicore disease, type I FSD myopathy, and myofibrillar myopathy.
Severity of CM is highly variable from patient to patient and within affected families. Infants with CM may have prenatal hypokinesia, deceptively low Apgar scores, and may be weaned from mechanical ventilatory support with great difficulty. Muscles with cranial nerve innervation are sometimes involved, resulting in ptosis, ophthalmoplegia, facial weakness, and dysphagia. Muscle contractures may be congenital or may develop during infancy, but they are usually not severe. In less-affected infants, modest motor progress may occur, but ambulation and acquisition of motor skills are delayed. Older children with CM usually are thin, have reduced muscle bulk and strength, and tend to avoid strenuous activity, but are able to perform ordinary tasks. Recurrent pneumonia or progressive scoliosis is frequent. Regardless of age at presentation, cerebral function is usually intact. Family studies have often identified minimally impaired or asymptomatic relatives with similar morphologic abnormalities in a muscle biopsy.
Although myotonic dystrophy is not in the strict sense a CM, central nervous system injury is unusually prevalent in affected infants (Figure 27-16) and may overshadow the natural history of the primary myopathy, the principal early manifestations of which are delayed muscle maturation and weakness (see Figure 27-8). Maternal weakness or fetal hypotonia may prolong labor. Myotonia in the mother is a helpful diagnostic sign.
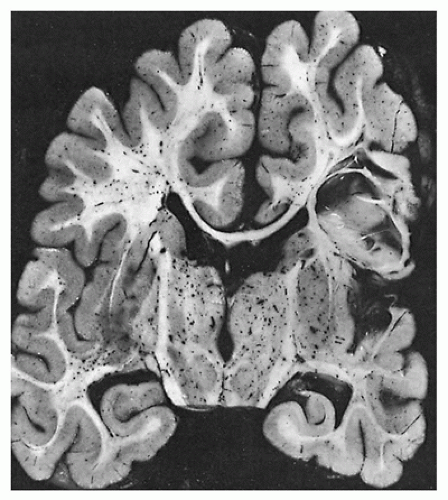
FIGURE 27-16 • Acquired porencephaly in a 2-year-old child who had symptomatic myotonic dystrophy with documented muscle maturation delay as an infant.
Identification and classification of the patient with CM is often challenging. Serum levels of creatine kinase, though typically normal, may be mildly elevated. Autopsy studies suggest that variation among muscles is common. Muscle biopsy, including electron microscopy, is essential. In samples lacking specific structural markers, the various subtypes of CM exhibit common features. Fatty replacement of muscle in CM may occur with age. Muscle fibrosis is exceptional in CM but may be observed in central core disease and in severe infantile nemaline myopathy
FIGURE 27-17 • A: Central nuclear and myotubular myopathy. Central nuclei are prominent and interstitium contains excess collagen. (Hematoxylin and eosin stain; original magnification ×300.) B: Central nuclear and myotubular myopathy. Myotubular configuration (arrow) is demonstrable in a variable percentage of fibers. (Methylene blue and azure II stain; original magnification ×800.)
CENTRONUCLEAR MYOPATHIES
Migration of reactive sarcolemmal nuclei away from the sarcolemma occurs in muscle regeneration and is a nonspecific alteration in chronic neuromuscular diseases of all types. Location of the nucleus in the exact center of most muscle fibers is the hallmark of centronuclear congenital myopathy (Figure 27-17). Sex-linked recessive, and autosomal dominant or recessive inheritance patterns of centronuclear myopathy are well documented (72,73,74,75). The autosomal recessive form is the most common. Severely affected infants are reported mainly in kindreds exhibiting an X-linked recessive inheritance pattern. Untypeable myotubes resembling the fetal myotube stage of development are prevalent in affected infants, but in contrast to the normal myotube, the perinuclear clear zone may extend for only a short distance on either side of the central nucleus. The clear zone contains glycogen, sarcoplasmic reticulum, mitochondria, and associated enzymes but lacks myofibrils. Overexpression of vimentin and desmin occurs only in the X-linked form (3). Mutations in the MTM1 gene that codes for myotubularin cause the X-linked disease (76). The dominant form is linked to the DNM2 gene, and the recessive form is linked to the BIN1 gene. Persistent myotubes uncommonly may be found in very young infants with other neuromuscular diseases such as myotonic dystrophy and type 1 spinal muscular atrophy.
FIGURE 27-18 • A: Nemaline myopathy, infantile form. Dark-stained, rodlike granules tend to be most prevalent in smaller muscle fibers in symptomatic infants. (Trichrome stain; original magnification ×300.) B: Nemaline myopathy, infantile form. Type I fibers are smaller than type II fibers. Smallest fibers are untypeable. (Myosin ATPase stain with preincubation at pH 4.3; original magnification ×400.)
Nemaline Myopathy
Nemaline (rod) myopathy (NM) is a distinctive and relatively common form of CM (77). Family data suggest two patterns of genetic expression: dominant with variable penetrance and autosomal recessive (78). Clinical subgroups include a rapidly fatal infantile form, a static or slowly progressive form with mild-to-moderate impairment, and a subclinical form in relatives. In most cases, the rods are readily observed in a cryostat section using a modification of the trichrome stain (Figure 27-18); rods are undetectable in sections stained with hematoxylin and eosin. Associated changes include type I fiber predominance and, in some instances, indistinct myosin ATPase fiber-typing reactions. Infants with nemaline myopathy usually have type I FSD.
Ultrastructural study of rods demonstrates discrete, electrondense bodies with characteristic crystalline substructures (Figure 27-19). In some infants with severe CM, rods are difficult to detect by light microscopy, but rodlike Z-band changes are prevalent in electron micrographs. Rods contain multiple proteins including a-actinin (a Z-band protein) and actin and are contiguous with Z-bands, aggregated beneath the sarcolemma or in some patients, located within the nucleus (79). Molecular studies indicate that mutations in nebulin and skeletal muscle alpha-actin cause the majority of cases of autosomal recessive NM (80). Mutations in several other NM-associated genes such as tropomyosin and troponin are uncommon. Nebulin mutations associate only with autosomal recessive NM; in other respects, genotype-phenotype correlation is poor and not useful for prognosis.
Only gold members can continue reading. Log In or Register to continue