- Cancer is a tale of two genomes: the patient’s original first edition and, derived from it but diverging over many years, the tumor’s evolving work in progress.
- Although much will be discussed about the role of unavoidable genetic risk factors and acquired mutations, we must not forget the powerful influence of modifiable factors. Estimates suggest that anywhere between 35 and 45% of all cancers may be strongly influenced by ill-advised lifestyle choices such as smoking, poor intake of fruit and vegetables, excess alcohol, lack of exercise, and development of obesity.
- Without wishing to kill the suspense, much of what follows will lead the reader inexorably to the following conclusions: that cancer cells are spawned but also shaped by experience; inheritance and environment conspire together to turn an outwardly normal conformist and idealistic cell into a cancerous and solipsistic pariah.
- The central dogma of molecular biology implies that information is transmitted as follows: the DNA codes for RNA (transcription) and then the RNA codes for protein production (translation). The process is deterministic, as each stage depends completely on information encoded within the DNA and RNA. Moreover, it is also believed to be essentially unidirectional, to quote Francis Crick, “Once information has passed into protein, it cannot get out again.”
- This view has required some revision since the 1950s. It has been known for some time that viruses can turn RNA into more RNA or even into DNA (hallmarks of RNA viruses and retroviruses). In fact, reverse transcription is routinely exploited in laboratory research. More recently, mammalian telomere maintenance and microRNA pathways provide further exceptions to the rule. Moreover, proteins can profoundly alter gene expression through epigenetic modifications such as methylation of DNA and acetylation of chromatin without altering the DNA code, though this falls rather short of being real reversal of information flow from protein to DNA. All of these processes have direct bearing on cancer biology.
- Any alterations to the usual flow of information can culminate in inadequate, excessive, or dysfunctional protein production and therefore in disease. Alterations to the DNA code, chromosomal rearrangements, changes in epigenetic regulation, and incorrect processing or translation of the mRNA can all disrupt normal protein production. Moreover, even subtle changes in the sequence or structure can drastically alter protein function and propensity to posttranslational modifications, both its own and that of interacting proteins.
- Cancers arise from normal cells by the accumulation of mutations or epigenetic changes (epimutations) that activate cancer-causing genes (oncogenes), inactivate cancer restraining genes (tumor suppressors), or facilitate the former by increasing DNA damage or compromising its repair (caretaker genes); mutations may be acquired by somatic cells or inherited through germline alterations in the DNA.
- Why do cancers happen in the face of all the various mechanisms designed to prevent it. Because DNA damage is unavoidable, cells affected by it innumerable and through replication cumulative – just throw in the key ingredients, nature and nurture and time.
- Currently, around 400 genes (or about 1 in 50 of our genes) are thought to be directly tumorigenic when mutated. Finding these can help unravel pathogenesis of a given cancer and could provide biomarkers to help define prognosis and even help select appropriate treatments and combinations. This knowledge is even being exploited in finding at-risk individuals before they develop cancer in order to target preventative treatments. Although there are some examples of all of these in clinical practice, there is still a long way to go before we can offer such personalized care for all.
- In some rare familial cancer syndromes, an essentially inactive copy of one of the key tumor suppressor or caretaker genes is inherited from one parent and confers a very high risk of developing cancer and often at a young age.
- In such cases, targeted screening for causative mutations in DNA, obtained by a simple blood test, is offered to other family members. This is exemplified by screening for BRCA1 and BRCA2 mutations in families with increased incidence of breast and ovarian cancers. However, the low incidence of mutations in these genes (less than 1 in 500 for BRCA genes) precludes their use in screening the general population. Genetic screening is also available for mutations in: the mismatch repair genes, MLH1 and MSH2, in some hereditary colon cancers; TP53 (Li–Fraumeni syndrome); RB1 (retinoblastoma); CDH1 hereditary diffuse gastric cancer; MEN (MEN1) and RET (MEN2).
- However, in most cases inherited susceptibility to cancer is determined by subtle variation in the coding and noncoding DNA of genes (polymorphisms, minor alterations in single nucleotides), each of which alone confers only a slight increase in risk of cancer. However, put a few of these together and mix in some carcinogens and you may well be several steps nearer to cancer.
- Genome-wide association studies (GWAS) that look for the presence of variation in genes by detecting single-nucleotide polymorphisms (SNPs) in large groups of patients have identified many susceptibility alleles for most common cancers. In general, the actual risk increase predicted by these is below 10% and has not translated into clinically useful screening tools to date.
- However, what does this 10% mean? Remember GWAS studies involve large numbers of patients. Does a given risk allele give everyone affected a slightly increased risk (10% closer to cancer, with the rest of the journey a chance affair driven by other inherited and somatic epimutations) or at the other extreme do 100 out of every 1000 individuals get cancer entirely because of it (i.e. this is very high risk for some people – we just don’t know who), or more likely somewhere in between?
- The mitochondrial DNA, inherited entirely from the mother, is also subject to mutations, and some conferring increased risk of prostate cancer, for example, have been identified.
- In most cases, however, epimutations are somatic and are not inherited from a parent but rather are acquired by individual adult cells. Within an evolving tumor, if advantageous to the cell such mutations will be propagated to an expanding clone of progeny cells by natural selection.
- Acquired mutations are tumor-specific and are not present in germ cells. Put simply, the patient’s underlying genome and that of the tumor will differ by the acquired mutations present in the latter. This fact is exploited in tissue-based biomarkers and in new treatments directed against those which are functionally important, one of the great success stories in modern cancer biology. Thus, pioneering successes in targeting the aberrant product of the BCR-ABL oncogene, in chronic myeloid leukaemia, with imatinib have been followed by other drugs directed against the constitutively active protein products of HER2 in breast and EGFR in lung cancer respectively. These successes herald a new era of personalized medicine, for these and other cancers, which many authorities now consider within our reach.
- A risk factor is anything that increases the risk of developing a particular disease and may be avoidable or unavoidable, modifiable or unmodifiable. Once described and risk determined, preventive therapies may be offered to those with one or more risk factors; or with suitable bombast, “what can be foretold may be forestalled.”
- However, in practice even effective and reliable preventative screening tests will not be implemented unless they are cost-effective, easily applied, and in themselves comparatively devoid of harm.
- The next best strategy is to diagnose the development of cancer at the earliest possible time so that curative treatments have the greatest chance of success. This in turn also requires the availability of reliable diagnostic tests, such as scans and biomarkers.
- Remember, biomarkers need not be functionally relevant to the disease process. In fact, the expression of any disease-irrelevant biomarker will do perfectly well so long as it recognises a cancer cell and may serve admirably as a shibboleth for disease classification and even in treatment selection.
- The ideal biomarker is one that can be detected in easily accessible body fluids that can be obtained with minimal invasiveness (e.g. saliva, sputum, urine, or peripheral blood) and would therefore be suitable for screening large numbers of people. Tumor cells (and other cells affected by the tumor) release proteins (and other molecules) into the circulation and these can be measured. Blood tests for prostate-specific antigen (PSA) (prostate), CEA (colon), CA-125 (ovary), and thryoglobulin (thyroid) are all in clinical use for monitoring patients after treatment and in screening for recurrence. However, their use as screening tools is limited due to poor sensitivity (they do not pick up early disease when curative surgery may be possible) and limited specificity (too many false positives). Even the best of these, PSA, is surrounded with controversy.
- Screening mammograms may be used for early diagnosis of breast cancer, a screening tool recently validated in large clinical studies.
- Geographical differences in cancer incidence, and the “migration effect” support the importance of environmental and lifestyle factors. Carcinogens are believed to act at least in part by promoting epimutations or by accelerating proliferation (and thereby propagation of cells with mutations).
- Smoking, diet, sex hormones, and increasing age influence risk of cancer. Recent studies have highlighted the risk of cancer in those with obesity and type 2 diabetes and that this risk might be reduced in part by use of metformin.
- Since groundbreaking cancer studies first showed that viruses can carry oncogenes and promote cancer by insertional mutagenesis, there have been major advances in the field of cancer-causing infections. Having fallen out of favor for some time, infection is again accepted as a significant contributor to up to 1 in 5 cancers globally – in particular the hepatitis viruses B and C, human papilloma virus (HPV), and the bacterium Helicobacter pylori are important causal agents in cancers of the liver, cervix/oropharynx, and stomach, respectively.
- Many countries have established vaccination programs for young women to prevent infection with cancer-causing strains of HPV to prevent cervical cancer. Other virus-related cancers are being actively studied. Various other organisms can contribute to tumor formation, with even the mysterious prion proteins implicated in pancreatic cancers.
Introduction
Heredity proposes and development disposes.
Sir Peter B. Medawar
’Tis misfortune that awakens ingenuity, or fortitude, or endurance, in hearts where these qualities had never come to life but for the circumstance which gave them a being.
William Makepeace Thackeray, The History of Henry Esmond
All cancers are caused by mutant genes. In some cases these may be inherited, but in most cases, cancer-causing mutations are acquired by non-germ cells (somatic cells) during life. Each cell experiences a unique set of environmental insults, internal malfunctions, and random setbacks. When added to a shared genetic makeup, this largely determines which cell will become a potential cancer-starter. No single cell lives through the same year as any other and to varying degrees they are exposed to a variety of DNA-damaging insults or make mistakes during the replication of their DNA. Together, the result is the emergence of cells with subtle differences in their DNA compared with the original inherited genome.
Where such mutations confer a growth advantage on a given cell, they allow it to give rise to an expanded clone and – by sequential acquisition and natural selection of further propitious mutations – to generate waves of clonal expansion culminating in the evolution of a cancer. Although the nature of these mutations varies from cancer cell to cancer cell, there are some common features. Thus, to date, just over 450 genes have been identified that may cause cancer when mutated. Of these, fewer than 20% have been shown to be inherited in the germline, and many of these appear to be involved in DNA damage responses and repair.
Inherited genetic susceptibility contributes to most if not all cancers in some way. In most cases, the cancers do not arise as a result of the powerful effects of rare mutations in single critical genes, though such cancer syndromes exist. Rather, in the majority of cancers genetic predisposition is a complex affair, operating through a range of less certain mutations and genetic variants that may increase risk of future cancer by a modest or small amount. In some cases, the risk associated with common allelic variations in peripherally relevant genes may be almost imperceptible, unless combined with multiple other gene variants or in the face of particular environmental challenges. Whereas mutations in genes acquired in somatic cells during life are the central players in cancer development and progression, inherited mutations may help to stage the process by increasing the available talent available for casting, thereby giving the whole performance a headstart on the path to cancer. This is exemplified by the earlier age of onset of common cancers in those with inherited susceptibility.
As this chapter traverses some of the most conceptually challenging terrain of cancer biology we will first try to provide the reader with an appropriate phrase book and travel guide to ease the journey. In time-honored tradition these aids will make some oversimplifications, which time spent among the chapters to come will slowly correct and embellish. By the conclusion of the chapter we hope to have given some insights into the role played in tumor initiation and evolution by genetic and environmental factors and in particular highlighted the extent to which susceptibility to cancer and responsiveness to treatment may be:
- predestined by genetic inheritance;
- influenced by environmental factors or infectious agents;
- determined by acquired somatic epimutations and natural selection.
To make sometimes difficult concepts more accessible we will on occasion take the scenic route and digress into a discussion of processes covered in depth elsewhere in the book. In some cases, such as the acquisition of genetic mutations (and epigenetic alterations) by single cells during adult life (as distinct from those inherited by all cells from the parents), this is unavoidable as it represents a major carcinogenic mechanism driven by environmental toxins and viruses. We also discuss how knowledge of these processes may actually pay dividends in the development of clinically important biomarkers and cancer screening tests for prevention, and early diagnosis of cancer and for use in a more functional subclassification of established cancers on the basis of predicted prognosis and response to particular treatments.
Cancer evolution as cellular snakes and ladders
Given the complexity of this chapter it will be helpful to bear this metaphor in mind while you read on. Cancer can be likened to a game of snakes and ladders. Viewed from the perspective of the many potential aspiring cancer cells that start as normal cells at the bottom left of life’s playing board, the ladders are oncogenes and the snakes are tumor suppressors. Those with inherited susceptibility start the game further up the board and nearer to the final square, where the cancer will be realized. Stem cells and cells exposed to mitogens throw the die more often and have a greater chance of landing on a ladder, but also on a snake. Hardly any cells make it to the end of the board, because this board is overpopulated with snakes. How do any cells make it? Only because tens of millions enter the competition and because the game runs for 70–80 years inside each of us. Yet, still only one in three of us will harbor a winning cell.
The Language of Cancer Genetics
Prophesy is a good line of business, but it is full of risks.
Mark Twain
Genes are runs of DNA that serve as blueprints for the manufacture of proteins – the functional and structural elements of the cell and of living organisms. It is known that less than 2% of genomic DNA encodes information for how a protein is put together, and another around 2% comprises regulatory regions that control how much, if any, protein will be made. This leaves a whopping 96% of DNA that, until recently, was largely, and as it turns out wrongly, dismissed as junk (more about this later).
With the exception of the sex chromosomes, you have two ostensibly identical copies of each chromosome and therefore two copies of each gene. Humans have 23 pairs of chromosomes – a pair of sex chromosomes (two X chromosomes in females and an X and a Y in males) and 22 pairs of autosomes. The number and configuration of chromosomes is referred to as the karyotype. Deviation from the normal karyotype is very typical of cancer cells and is referred to as aneuploidy. Genes usually occupy the same position on a given chromosome (the genetic locus) in all individuals unless something has gone badly wrong with the karyotype (note the Philadelphia chromosome in chronic myeloid leukemia and the key role of translocations as a contributory cause to cancer-relevant gene mutations). This does not mean that the two genetic loci on the two chromosome copies are necessarily completely identical.
Genes occur naturally in different versions (different nucleotide sequences), which may or may not result in the variants conveying different information, and these variants are referred to as alleles. Broadly, those alleles which occur at a frequency of greater than 1% in a population are termed polymorphic and those variants which occur at below 1% are termed subpolymorphic. To put this in context, most highly penetrant gene mutations (ones that almost always produce an unmistakable and clinically unpleasant outcome) occur in less than 0.1%.
In general, the genes you have in all their various allelic variants is your genotype; the physical manifestations of your genotype (how you look, function and what proteins you make) is your phenotype. Much of modern biology, and cancer biology in particular, is concerned with describing the effects of variations in genotype on resultant phenotype. Many mutations may not have a discernible effect on phenotype at all, or may indeed occasionally even improve the function of the protein product and, as a result, the overall viability of the organism in a particular environment – the “propellant of evolution.” As first predicted by Charles Darwin, advantageous mutations will be retained and become increasingly common in a population, whereas disadvantageous ones will be eliminated through natural selection. This is because the lucky organisms with the new, better gene outcompete those with last year’s model and raise more offspring, some of whom will have the better version and will themselves have more offspring and so on. Remember that natural selection is critically determined by the environmental context in which this takes place – lots of thick hair is useful in the cold but not when it is 40 degrees in the shade.
If you have one gene altered from the norm in some way you are heterozygous for that alteration (two different alleles); if both genes are unaltered or both similarly altered this is homozygous. Confusion sometimes arises when different mutations in the two alleles cause the same phenotype (e.g. neither gene makes any viable protein). In such cases the terminology will reflect the level of scrutiny that has been employed, whether this is based on phenotype observation or on sequencing of the genomic DNA. Please note that new alleles or mutations are only passed on to future generations of the organism if these are present in the germline – in sperms or oocytes or their precursors. On the other hand, mutations in somatic cells can be transmitted to cellular progeny during replication – in fact this is the predominant driver of tumorigenesis, ultimately giving rise to a clone of cells with a common ancestry – but such mutations are not passed on to the offspring of the organism.
Another important concept often referred to in the book, is that of highly conserved genes. Such genes appear in near-identical forms across many species and in general do not naturally occur in multiple alleles. The implication is that such genes have been around for a very long time and have long ago reached “Nirvana.” The “Über”-proteins so made are the “Mary Poppins” of proteins – practically perfect in every way. Thus, any mutations will result in an inferior protein and will be eliminated over time.
On the other hand, genes that occur in several allelic variants of significant frequency in a particular population are not necessarily still a “work in progress.” Recent mixing of different populations, each of which has selected the best alleles for their previous environmental context, can increase genetic variation. This is mostly a good thing because it reduces chances of nasty recessive diseases. However, matching the wrong alleles to the wrong environment can also potentially increase risk of diseases such as diabetes and some cancers if, for example, alleles best suited to low-energy diets are placed in an environment replete with energy-rich foods, either because the diet has been imported or the alleles exported!
Before we become over-enamored of our Darwinian view of cancer evolution, we should first set out the unique parameters within which this analogy holds water. In contrast to the evolutionary history of life on this planet, the whole dynastic descent of the cancer cell is played out within the space of a single human lifetime. With the dubious exception of fragments of cancer DNA pollinated by viruses, the death of the host signals the death of the cancer; the peculiar afterlife of the HeLa cancer cell line cannot in all honesty be regarded as their transcendence over Henrietta Lacks. Thus the game may follow a set of similar rules but the prize in the case of cancer is not ascendency but oblivion; there can be no clearer example of the essential difference between competing species and cancer cells, however shared the propelling force of mutations and natural selection. The latter evolve against a backdrop of cellular cooperation and selfless mutual support with other cells, because at least in part they are all closely related members of the same family – they are neither finches nor cuckoos, simply the black sheep of the family.
Cause and Effect
In the strict formulation of the law of causality – if we know the present, we can calculate the future – it is not the conclusion that is wrong but the premise.
Werner Heisenberg
In most cases, the effect of any single allele appears to be very small and it is likely that discernible phenotype alteration is a product of allelic variation at many different genetic loci. Not all genotype–environment interactions necessarily change for the worse. The Japanese normally have greatly increased risks of stomach cancer compared to North Americans, but within a single generation those who emigrate to North America avoid this risk and instead experience that of the indigenous population, probably due to dietary changes. (The risks of diabetes and coronary disease show the opposite trend!) It is also worth noting that deciding to what extent these effects result from environment alone or gene–environment interactions is often challenging. Note how relatively few North Americans move to Japan and adopt that native lifestyle and diet, thereby precluding a test of increased gastric cancer risk with that direction of travel. It is often assumed that environmental factors and allelic variation may operate in the pathoetiology of the great majority of cancers. However, as we will see, in some cases genetic or environmental factors alone may be the major driving force.
At one extreme, certain genes are very important and must be in good working order for a long and healthy life (high-risk genes). Alterations in these important and often highly conserved genes almost invariably end in a very evident and very bad clinical outcome (high-penetrance mutations) and diseases resulting from such inherited mutations generally occur at a younger age than the sporadic counterpart. To keep things relevant, such inherited mutations may result in a complete failure of that gene to direct production of a cancer-restraining protein, or a protein product is dangerously changed or activated in a way that promotes cancer. Such changes give “would-be” cancers a headstart, explaining why these cancers often present at a younger age than the more usual sporadic version. These monogenic diseases (the alleles of a single gene determine the presence or absence of disease) are termed either dominant or recessive, depending on whether they require one or both inherited copies of the gene to be mutated. In cancer this is a bit more complex than for other inherited diseases because we have to worry not just about inheriting bad alleles in the germline, but also about somatic gene alterations (mutations) occurring in adult life in individual cells. Remember that cancer can uniquely start with genetic alterations in just one cell.
For those genes that promote cancer when activated, oncogenes, a mutation in a single copy may suffice, whereas for a gene which puts the brakes on cancer, a tumor suppressor, both gene copies must be inactivated. But, because life is complicated, there is also a half-way house in which a slightly less bad outcome may follow loss of a single gene (for example the reduced protein dose means you still have an increased risk of cancer but at an older age) – such genes are termed haploinsufficient.
Big Changes and Big Consequences
Hereditary cancers resulting from major genetic defects are rare, but such families have been extensively studied and much has been learned about cancer biology as a result. Generally such hereditary cancers follow Mendelian principles and are readily diagnosed if affected family members are known. OK, if this is all true then why do the best-known familial cancers involve inheriting a single nonfunctioning copy of a tumor suppressor gene such as RB? There are two explanations for this example: (1) Because there are so many cells with only one working copy of the tumor suppressor and so many opportunities to acquire mutations during adult life, it is inevitable that the good copy will get lost in one cell or another (the two-hit hypothesis) and set the scene for tumorigenesis. (2) The tumor suppressor is haploinsufficient (you need all the protein you can make, you have none to spare, and half as much isn’t enough to prevent disease).
It may be helpful here to consider these two scenarios in traditional terminology. In scenario 1, cancer is autosomal dominant at the whole organism level, as pretty much everybody with inherited loss of RB will get cancer, however when we start to look at the genome at a cellular level this is autosomal recessive as both copies of RB need to be inactivated. In scenario 2, the disease may appear autosomal dominant but there will be a dose-effect with generally worse outcome if both copies are lost. Over the last 30 years there have been numerous successes in finding high-penetrance susceptibility genes for cancer, including the mismatch repair genes in Lynch syndrome–related colorectal cancer and BRCA genes for breast and ovarian cancer. However, in each case these genes account for less than 90% and 80% of the hereditary risk of these cancers, respectively. At the time of going to press, a further high-penetrance susceptibility gene for ovarian cancer, RAD51D, was identified.
Small Changes and Butterfly Wings
Much more common and more difficult to pin down are the effects of more subtle variation (low- to moderate-penetrance mutations) in one or multiple genes. As mentioned, many genes occur naturally in subtly different versions, which all still work more or less and do not cause inevitable and easily observed changes in the individual. Thus variant genes (alleles) may result in proteins that differ by as little as one amino acid, may have no discernible effect whatsoever either on protein function or on disease susceptibility or might, like the flap of a butterflies wings, conjure up a cancer in the future. Implicit in this view of risk is the subordination of simple random bad luck. In other words, we erroneously accept as fate what we lack sufficient information and understanding to have predicted. If a seemingly miniscule change can alter the course of a life, then we only need to detect those changes or conversely introduce small ones of our own design and a satisfactory outcome is guaranteed.
However, as with most of life’s tribulations, responsibility rarely rests with any single factor but rather a series of events taking place in the wrong place at the wrong time may result in catastrophe; given the complex chain of happenstance it is understandable that we may often fail to identify it. Thus, even comparatively weak effects are amplified and given opportunities for fortuitous synergies with other mutations over time; this is compounded by the pervasiveness of hereditary mutations and allelic variants that affect all somatic cells from the off, and of those acquired ones propagated by clonal expansion during life. Moreover, if tiny changes can have drastic consequences over long periods of time, then we must have tools sufficiently sensitive to measure these tiny changes accurately enough, or the resources to prospectively follow the progress of patients for long enough to make the effect obvious.
This is no minor issue, as the inability to measure precisely enough limits our ability to make predictions about the future behavior of many complex systems. Weather forecasts – as an oft-cited example – offer a tolerable level of accuracy only over a week at most. In forecasting cancer, what chance do we have to determine every single relevant genetic, epigenetic, and other variation in all individuals at the outset and thence all the potential somatic variations accumulating during life? Don’t get despondent though, because even if we accept that complete understanding may never be granted us, we have made, are making, and will continue to make progress in finding risk factors that we can do something about. Moving beyond politics-inspired rhetoric, progress has been made in linking allelic variants with disease risk, and some notable examples will be discussed later. All that was needed were sufficiently large numbers of individuals who could be genotyped by various means and then followed up until a detectable pathology developed in some.
There are practical ramifications to this discussion, if many cancers arise just through simple bad luck then we should stop agitating over the future and get on with life. However, if as seems likely, it is a blend of predictable and modifiable factors that may measurably increase risk and then some bad luck thrown in, then we can successfully adapt. The challenges are substantial. Even with hindsight the causes are often difficult to pin down and it is tempting to apportion blame to the most evident or prominent among these, even if this proves intractable as a prevention target, when a more nuanced understanding might have revealed a readily avoided or treated risk factor.
Predicting the Future
Physic, for the most part, is nothing else but the substitute of exercise and temperance.
Joseph Addison
Can we state this in molecular terms? Much of the hope in personalized medicine is predicated on a belief that by determining variations in multiple genes we may be able to make fairly accurate predictions about the future experiences of disease in later life and help guide individuals towards avoiding risky behaviors and even preventative medicine. Where does random chance fit in? Try as we might to exorcise chance events from our world, it is almost certain that many of the mutations underlying disease susceptibility arise by chance events or due to unavoidable insults – so for now the difference may have no practical significance. But perhaps the single most random event in determining genotype is sexual reproduction – self-evidently you have no opportunity for selecting your parents and it is certain that they exercised little logic in selecting one another.
Let us stray briefly to consider some clinical genetics scenarios that exemplify some of these concepts. Familial hypercholesterolemia (FH) is the most common monogenic disease and is caused by a high-penetrance (high-risk) mutation that affects 1 in 500 people. It is autosomal dominant, so that on average one out of every two children born to an affected individual will also get FH and as a result will develop the very high cholesterol that typifies the disorder – so far entirely predictable. However, even if untreated, not all individuals with FH will die early through cholesterol-related complications, mainly heart attacks, though most will. Conversely, not all FH patients who are effectively treated will avoid an early heart attack, though again most will. Why not? Because variations in many many other genes, known and unknown, influence the extent of the phenotype, and others affect the risk of heart disease and then we have the influence of lifestyle thrown into the brew. Together, these can be positive and potentially offset the risk posed by a high-penetrance mutation or, on the other hand, negative and negate the benefits of cholesterol-lowering treatments.
This is important to bear in mind because one can easily become seduced by the simple view of biology derived from studying cells and animal models. Such models, although extraordinarily useful in addressing specific questions, have certain limitations. Thus, unlike humans, inbred rodent models of diseases such as FH or of cancer are very similar genetically (and inhabit a uniform, controlled, and sterile environment) and therefore if you can cause a cancer by a particular genetic manipulation(s) in one it will hold true for all, whereas this is manifestly not the case for humans, in whom genetic and lifestyle variation will powerfully modulate the effects of any high-penetrance mutations (both positively or negatively). The importance of looking at genes in functional interacting networks is likely to be even more critical when the effects on cancer risk of any single variant gene are going to be slight, particularly where there are no readily measured metabolic or physiological consequences, as is mostly the case with cancer risk. Moreover, as the variant gene may only have an effect, for example, in the presence of some external agent such as tobacco smoke, or when following a bad diet, the immensity of the task is only too apparent.
Genome-wide association studies (GWAS) are now increasingly identifying associations between common variability and cancer and have revealed, perhaps unsurprisingly, that common genetic variability does not explain all of the genetic predisposition to disease (see Box 3.1). Advances in technology as well as reduction in costs are allowing some of the apparent gap to be addressed by genome sequencing and data processing (see Chapter 20). However, huge numbers of patients may have to be studied and at great cost financially and in time in order to identify these relatively low-risk genetic susceptibilities.

Published genome-wide associations through December 2010, 1212 published GWA at p ≤ 5 × 10−8 for 210 traits. Taken from the National Institutes of Health GWAS website (www.genome.gov/gwastudies/). Credit: Darryl Leja and Teri Manolio.

Again, a clinical example here may be helpful and encouraging. Even where the disease etiology is clouded and seems to involve a plethora of complicated interacting genetic variants and lifestyle, we can take positive and useful action. In the case of type 2 diabetes, disease may only arise if at some point an at-risk individual makes a lifestyle choice ill-suited to their specific genotype (whatever that may be); thus, sloth and gluttony are particularly ill-suited to somebody with a strong family history of type 2 diabetes. Why? Because, with a 1 in 2 chance of developing diabetes the odds of surviving such a lifestyle would be stacked against you. We do not need to know anything about genotype before recommending a healthy lifestyle and avoiding obesity. It would, however, be useful to have some biomarkers that narrow down risk further, and many studies are addressing this.
Nonhereditary Mutations
There is, of course, another way in which altered genes come about. Genes can also become altered in single cells during adult life – somatic mutations – under the influence of chance events during DNA synthesis, byproducts of oxidative metabolism, or through exposure to carcinogens. And, of course, to complicate things, this may happen because of mutations in genes that otherwise help you to avoid acquiring mutations (exemplified by genes encoding proteins needed for DNA repair). Genes may even become silenced by changes in methylation without any alterations in the DNA code (see Chapter 11). These factors all represent challenges for those attempting to find biomarkers, because:
- such changes are confined to affected cells within the tumor or proto-tumor, which may be undetected or inaccessible;
- even if cells or cell contents find their way into more accessible body fluids it may no longer be possible to quantify cancer-related proteins or mRNAs reliably and epigenetic changes will not be picked up by screening for mutations in circulating tumor cells;
- these somatic changes are essentially restricted to the individual patient and will not be inherited by any offspring, though the propensity to acquire such changes could be hereditary!
A Light at the End of the Tunnel
Despite the complexity of the task, there have been notable successes in cancer risk prediction and genetics. Obesity and resultant sequelae such as diabetes greatly increase risk of certain common cancers and are fairly readily screened for and preventive strategies for obesity are being developed in many countries. BRCA genes have shown their value as predictors of risk of breast and ovarian cancer, the presence of cancer-specific mutations in growth factors or estrogen receptors is now used to guide appropriate targeted treatment in breast cancer (herceptin or anti-estrogens), and, despite recent controversies, prostate-specific antigen is in widespread use as a screen for prostate cancer. A more detailed discussion of the practicalities of risk and risk screening follows below.
Risk Factors
“Winwood Reade is good upon the subject,” said Holmes. “He remarks that, while the individual man is an insoluble puzzle, in the aggregate he becomes a mathematical certainty.”
Sir Arthur Conan Doyle, The Sign of the Four
A risk factor is anything that increases the likelihood (probability) of an individual developing a particular disease, such as cancer. Some risk factors are modifiable by the patient, such as tobacco and saturated fat consumption, some, such as diabetes, may be modifiable by the use of therapeutics. On the other hand, some risk factors, such as age, family history, and gender are simply not modifiable, though the risk of cancers associated with these can be reduced by targeting other risk factors that are themselves modifiable. Importantly, not everybody with risk factors gets cancer.
It is worth pausing here for a moment of quiet reflection. Risk is a difficult concept for most clinicians, and even more so for most patients. Professionals are often best at predicting what has become obvious to them, but is not necessarily so to a patient. For example, radiological studies prompted by a persistent cough may subsequently demonstrate metastatic lung cancer that will almost invariably drastically shorten life, even with most current treatments. A prognosis offered under these circumstances has considerable individual validity, and patients and carers may plan for the future with some degree of certainty. At the other extreme, however, the presence of certain risk alleles may well show a statistical association with future risk of developing a particular type of cancer, but this may be very slight and only manifest in those who pursue a particular lifestyle. In practical terms, the low risk means that individualizing any risk-modifying strategy, even one proven effective in clinical trials, is fraught and will not necessarily benefit all. Some will live a bit longer, some may live a lot longer, and some not at all – and no one will ever know which patients did what. How do you advise in such a case? It is often worth considering the concept of avoiding risk using simple analogies.
Drowning is clearly a bad thing which we should endeavor to prevent. But how? Do we offer risk-reducing strategies to everybody – or focus on those above an agreed threshold of risk? People who live near the sea are more likely to die of drowning than a Bedouin in the Sahara Desert. Wearing of a lifejacket might well be a cost-effective and accepted strategy for the former but would be wholly inappropriate for the latter. We could try and individualize the intervention more accurately by determining other risk factors, such as ability to swim and actual exposure to risk activities, such as sailing or swimming. Finally, we should have some idea of whether the intervention is devoid of harm. Making sure that everybody can swim would be a reasonable public health approach and would perhaps have additional benefits, such as fitness, and could be rolled out across whole populations. Stopping people swimming or sailing completely would be inappropriate as it would contribute to obesity, might compromise livelihoods, and would have a significant negative effect on quality of life.
As far as lifejackets are concerned, nobody would be convinced to wear one if they were never exposed to bodies of water they could ever drown in. This is not academic, as any risk-avoidance advice we give will inevitably carry with it some adverse socioeconomic or health consequences. There are people now whose lives have been blighted by the knowledge that they are at risk of a particular cancer in the future. The anxiety may be worth it if an effective preventative therapy exists, but the relative risk–benefit must be assessed (see Chapter 16). It is also important to note that even where risk of a particular cancer is strongly related to the presence of a nonmodifiable risk factor, such as female gender and breast cancer, risk may be successfully modified by targeting something else, such as weight and use of exogenous estrogens, for example.
Bradford Hill and Modern Epidemiology
In clinical practice, how do we identify a potential causal relationship between an identified factor and a disease? In the 1960s, the British statistician Sir Austin Bradford Hill formulated a set of minimal conditions to provide evidence for causation. These criteria are as follows:
- Temporal relationship (the cause has to precede the effect)
- Strength of association (statistical tests of the relative risk associated with the factor)
- Reproducibility (the same relationship is consitent and replicated across different studies)
- Dose–response relationship (greater exposure means greater risk)
- Modifiable by experimentation (linked to above – intervention to minimize exposure reduces risk)
- Plausibility (consistent with known biological or pathological understanding)
- Coherence (within the context of overall knowledge – may be broader than plausibility)
- Specificity (a cause produces a specific effect, though this may be difficult to apply to risk factors that damage more than one tissue)
- Analogy (are there other similar examples and have other explanations been considered and tested by experimentation etc).
A risk factor for cancer is just that – its presence does not invariably guarantee that an individual will go on to develop a particular cancer nor does its absence imply that the person will not. No matter how comforting the notion, a butterfly flapping its wings will not always conjure up a storm and neither will a host of frenzied eagles. Thus, being a man is a risk factor for prostate or testicular cancer but not all men will be affected. Obviously, some risk factors are much more predictive than others, but those known to predict a very high risk of developing cancer are relatively few. The best-known risk factors are not modifiable and the most important is age. The older you are, the more time you have had to acquire cancer-causing mutations and to be exposed to environmental carcinogens. Incidentally, under most circumstances gender is not regarded as a modifiable risk factor, but some aspects of gender, such as hormonal profile, are.
In terms of avoidable or modifiable risk factors, the greatest successes have resulted from the screening of whole populations or genders for signs of developing cancer or by identifying strong and avoidable or treatable environmental risk factors such as tobacco smoke, various occupational hazards (e.g. asbestos exposure), obesity, and infection with viruses (e.g. HPV). The relevant action has then proved relatively straightforward – don’t smoke, follow safe working and sexual practices, eat well and exercise, accept vaccination against HPV if you are a young woman and attend for screening cytology or radiology after a certain age.
Finding the occult vulnerable before they get cancer or at least while they are still in the early curable stages of the disease is now the big challenge – that is, finding all those many people out there who are at increased risk of cancer for some as yet unknown or unsuspected reason. These groups can then be offered preventative treatments and more rigorous screening. This will require a molecular and/or genetic approach, which has to date proved much less successful. In fact, there are remarkably few reliable screening tests able to accurately identify those at risk of particular common non-inherited (sporadic) cancers, and even fewer that will accurately foretell those who will experience serious adverse effects or early death as a result of that cancer (an issue exemplified by recent controversies surrounding screening for prostate cancer in men). Actually, this becomes a matter of philosophy – determinists hold that the universe proceeds through a chain of events conforming to the laws of cause and effect.
By implication, finding risk factors that do accurately predict the future development of cancer might eventually allow individuals or particular groups of individuals at high risk of developing cancer to be identified, opening up the possibility of preventative medicine. Such an approach, involving screening of individuals for cardiovascular risk factors in order to most effectively target preventative drug treatments, has prevented heart attacks in those who already have coronary disease (secondary prevention) and in those with diabetes, adverse family histories, and even in unselected apparently healthy individuals (primary prevention). However, this has been achieved largely because high-risk groups are readily identified and prioritized for screening and subsequently cardiovascular risk factors are readily determined in the clinic by recording blood pressure, age, gender, history of smoking, family history of heart disease, and taking a blood test to measure cholesterol. Moreover, extensive epidemiological and prospective studies of huge numbers of patients have allowed us to create reasonably accurate and effective risk charts that relate all these parameters to future risk of heart attack. Finally, many of the main risk factors are readily modifiable by drugs that are themselves remarkably free of potentially harmful side effects and are of proven benefit in large trials.
For cancer, few established risk factors are currently available and they are often fairly nonspecific. Thus, all women are currently regarded as at risk of cervical and breast cancer, and screening is offered to all after a certain age (see below). All smokers are potentially at risk of lung cancer and all are encouraged to give up, but no specific screening programs are established, though one could envisage some form of regular lung imaging in smokers to try and find cancer at an early and potentially curable stage. Tests that may identify higher than average risk groups within these populations are in development but are yet to become widely used and are far from perfect. In the case of cervical screening, universal uptake of HPV vaccination may render improved screening redundant. Similarly, the increased risk of cancer in obese and diabetic patients may be managed by improving lifestyle and by application of drugs such as metformin, rather than by developing better biomarkers or actively looking for cancer. Other clinically apparent risk factors that are currently used to target at-risk groups for cancer screening, include the presence of multiple colonic polyps or ulcerative colitis, but as the screening tests then employed are designed to diagnose cancer early they are not strictly preventative. What is immediately obvious from this list is the near absence of measures of clinically silent risk factors that predict the future development of cancer (there are as yet few parallels for serum cholesterol or blood pressure readings). Finally, preventative treatments, aside from lifestyle modification, are generally similar to those used to treat established cancers and are expensive and often highly toxic.
Lifestyle could realistically be targeted without any form of screening on the basis that good diet and exercise should be good for all. Many studies have reported on the substantial increased risk of breast and colon cancer in obese people and those with type 2 diabetes. Unfortunately, in the United Kingdom, people with a healthy body weight are becoming rarer. Two in three men (67%) and more than one in two women (56%) are overweight or frankly obese, and this is increasing at an alarming rate. If current trends continue, we will all be obese by 2050! Some encouraging results suggest that some of the risk of cancer can be offset by treatment with metformin. Sadly, achieving changes in lifestyle has proved more difficult.What about less well-known lifestyle factors potentially posing a risk of cancer? Recently, the EPIC (European Prospective Investigation into Cancer and Nutrition) study has started reporting on dietary influences on cancer risk in (more than 500 000 people in Europe, including close to 100 000 in the United Kingdom).
Developing screening tests for primary prevention of cancer is clearly the ideal, but screening for recurrence or progression in those with established cancers is also important. There are around 2 million cancer survivors in the United Kingdom (2008 estimate) with breast and prostate making up the largest groups. Moreover, these figures are increasing at around 3% year on year. So this is a very sizable task in itself. Most current useful biomarkers are those such as PSA, CEA, and CA-125 that are used to monitor cancers of prostate, colon, and ovary post treatment, respectively.
In this section we will discuss recent progress in addressing these issues and also discuss what is known about the relative roles of nature and nurture in the causality of cancer. Recent rapid progress in genotyping of individuals as a potential means of predicting at risk groups for various cancers and in molecular profiling of tumors (including sequencing tumor DNA to find mutations, and looking at changes in gene and protein expression) to tailor treatments and predict outcome will also be addressed. The current status of various carcinogens and cancer-causing organisms is also discussed.
It is easy to come away with the view that our lives are spent entirely in pursuing suicidal lifestyles in a world overflowing with cancer-causing agents. This is not the case. Maybe we should, on this occasion, be guided by someone who is – arguably – not one of history’s most risk-averse characters.
The torment of precautions often exceeds the dangers to be avoided. It is sometimes better to abandon one’s self to destiny.
Napoleon Bonaparte
Preventing Cancers
She could not explain in so many words, but she felt that those who prepare for all the emergencies of life beforehand may equip themselves at the expense of joy.
E.M. Forster, Howards End
Although there have been some notable successes in cancer prevention, including screening for breast and cervical cancer, readily determined risk factors for future development of cancer are generally not available for the common cancers. In fact, we must remember that in many cases we have no idea why a particular person has developed cancer, even if we assume the operation of the various mechanisms described in this book. However, as has become clear already, in some cases risk factors have been identified and this knowledge has helped us to develop some specific preventative strategies, ranging from public health initiatives aimed at the whole or a large part of the population through to genetic testing offered to selected individual patients with a worrying family history.
Most tests for potential “markers” of risk or of early disease are complex and invasive or require costly imaging that is not always universally applicable (cervical cytology, biopsy, X-ray, and scanning). Moreover, these largely pick up established disease, whereas the ideal screening test would identify individuals who have as yet not developed cancer at all. There are several examples of screening tests (see Chapter 18) that will be discussed later and in Chapter 17. Even where noninvasive markers that precede cancer onset have been described, the predictive strength is often not robust enough to guide use of preventative treatments, particularly where these are potentially harmful, such as surgery. Currently, mastectomy and salpingo-oophorectomy are widely used by carriers of BRCA1 or BRCA2 mutations to reduce risk of developing tumors of the breast and ovary, respectively, but how many would never have developed clinically meaningful cancers without such surgery is not clear.
One approach to improving the accuracy of predicting the future risk of cancer in individuals is to look at more than one risk factor concurrently (as is done for heart disease), many of which have been and continue to be described. It is also hoped that information on heritable genetic factors in the patient, as well as on how individual genes may become mutated specifically in cancer cells, may also help in prognostic and treatment decisions.
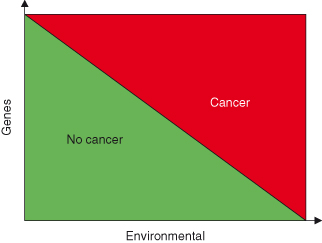
Risk factors may be environmental (lifestyle-related or unavoidable) or genetic (inherited) and are generally specific for particular cancers. Inherited factors are clearly of major importance and an historical overview of theories of heredity is given in Box 3.2. However, most cancers arise through a combination of genetic and environmental factors and less commonly by extreme exposure to radiation, viruses or cancer-causing chemicals (carcinogens) or combinations of genetic factors only (Fig. 3.1). Having a risk factor, or even several, does not guarantee that an individual will get the disease, but such guarantees rarely exist in disease prevention (where hindsight is not yet available!) and are unlikely to do so in the foreseeable future. Hence the attraction of early diagnosis, where the disease is at least present (even if outcome is less certain). What is practically required is a means of calculating risk of future cancer and cancer-related morbidity or mortality accurately enough to be able to safely apply preventative treatments. In other words, if that treatment is an entirely nontoxic and cheap drug, then treating a few individuals who would never have developed or suffered as a consequence of disease is OK, whereas if the treatment is mastectomy it is not (see Box 1.1 – Screening).

Figure 3.1 Cancer susceptibility is a result of a combination of genetic and environmental factors. The cumulative effect of both genetic and epigenetic changes and exposure to environmental factors determine the likelihood of developing cancer. In this model, only in extreme cases will cancers arise entirely due to either genetic factors alone or environmental factors alone.
Many risk factors for cancer are avoidable. There is no need to screen individuals before advocating smoking cessation, healthy diet, and using sunscreen, for example. Such recommendations are public health issues and should apply to all. It was 50 years ago that Richard Doll and A. Bradford Hill reported findings on a cohort of British doctors showing a strong link between cigarette smoking and lung cancer.
Risk factors and cancers can broadly be divided into two categories:
Analyzing cancer genomes can contribute to disease subclassification, improved prognosis, and even treatment selection, and many genetic variants are now employed as biomarkers in these areas. Different genomics techniques including those to determine SNPs, gene rearrangements, alterations in DNA copy number, epigenetic changes, and differential gene expression have all proved useful in this context.
Cancer Genetics – in Depth
You know how often the turning down this street or that, the accepting or rejecting of an invitation, may deflect the whole current of our lives into some other channel. Are we mere leaves, fluttered hither and thither by the wind, or are we rather, with every conviction that we are free agents, carried steadily along to a definite and pre-determined end?
Sir Arthur Conan Doyle
Mutations
As natural selection works solely by and for the good of each being, all corporeal and mental endowments will tend to progress towards perfection.
Charles Darwin
The term “mutation” can refer to any type of change in DNA. Exposure to genotoxic carcinogens can result in various differing forms of mutation. However, it is important to remember that mutations can also occur under the influence of unavoidable DNA damage, such as that induced by oxidative stress, background or cosmic radiation, and also by simple errors arising during DNA replication. The process by which proteins are made – translation – is based on the “reading” of mRNA that was produced via the process of transcription. Any changes to the DNA that encodes a gene will lead to an alteration of the mRNA produced. In turn, the altered mRNA may lead to the production of a protein that no longer functions properly. Even changing a single nucleotide along the DNA of a gene may lead to a completely nonfunctional protein. Mutations in one or more genes can therefore lead to disease.
The genetic changes that lead to unregulated cell growth may be acquired in two different ways – they can be inherited or they can develop in somatic cells. The phenotype of cancer cells result from mutations in key regulatory genes. The cells become progressively more abnormal as more genes become damaged, particularly when the genes that regulate DNA repair and checkpoints become damaged (see Chapters 7 and 10). Most cancers are thought to arise from a single mutant precursor cell (in other words they are “clonal”), with further clones originating by accumulation of further mutations, and those clones that gain a growth advantage will tend to take over the population (clonal expansion).
One aspect of this view of cancer is that the transition from a normal, healthy cell to a cancer cell occurs via the stepwise accumulation of mutations in multiple different oncogenes, tumor suppressor, or caretaker genes (Fig. 3.2). This model also accounts for the prevalence of cancer particularly in older individuals. Although the number, identity, and order in which mutations occur will likely vary enormously between individuals and different cancer types, attempts have been made to quantify the likely number of mutations required to generate a transformed human cell in culture. Studies from the laboratory of Robert Weinberg and others support the view of cancer formation as a multistage process, as suggested by Armitage and Doll in the 1950s, by demonstrating that at least 4–6 interlocking mutations may be needed to transform cultured human primary cells. However, whether this also equates to the requirements for formation of all cancers in the context of the intact organism remains controversial. Controversy remains because as few as two interlocking mutations may suffice to generate cancers in rodent models, a hypothesis that is difficult to test in humans, as the very earliest stages of cancer are generally not available to the researcher.
Figure 3.2 Multistage carcinogenesis – the concept of initiation and promotion. Genetic changes induced by mutagens are irreversible but may be phenotypically occult until further events such as proliferation or loss of differentiation unmask them. The mutagen or carcinogen is the “tumor initiator,” but other factors (tumor promoters) affect whether mutated cells proliferate and form tumors. Promoters can contribute to cancer formation but do not alter DNA. Promoters increase the frequency of tumor formation in tissues previously exposed to a mutagen or tumor initiator. For example, skin papillomas form after exposure to carcinogens, but there may be considerable latency between the mutation in the stem cell pool and exposure to a tumor promoter that promotes proliferation, resulting in a visible lesion. This expanded clone will then itself be vulnerable to subsequent mutational events (hits) for tumor progression. Some evidence suggests that angiogenesis may accompany the growth of the tumor, or it may arise through a distinct mutational event (angiogenic switch). Finally, an invasive cancer forms with cells entering the circulation. However, it is generally believed that a further mutational event (such as inactivation of a “metastases suppressor” gene) is needed for colonies of cancer cells to establish in a distant location.
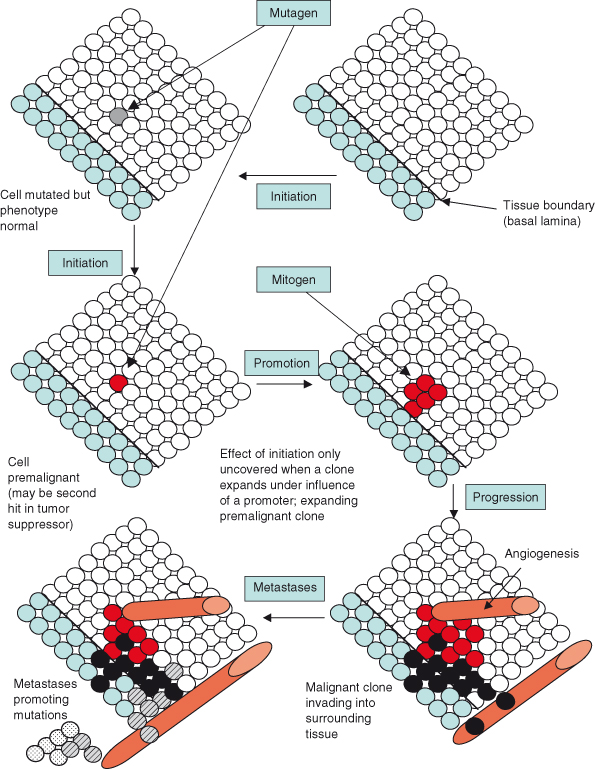
It is possible that intrinsic differences between human and mouse may be important and these might include the relatively longer telomeres found in mouse cells for example. However, it is also possible that the larger number of mutations required to generate a transformed cell in vitro than required for a cancer in vivo, might be explained by the absence of the potentially supportive effects of environmental factors extrinsic to the cancer cell, such as the tissue location, stroma, and vasculature from transformation assays in vitro. Defining the minimal platform for oncogenesis in vivo is likely to be an important and interesting area for study over the next decade.
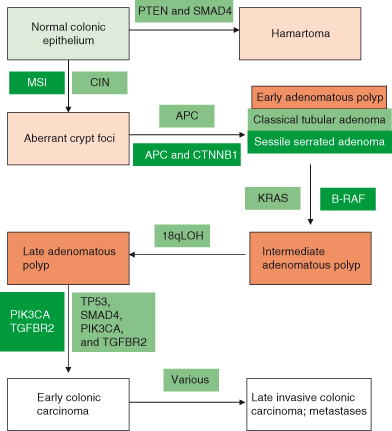
The actual number required notwithstanding, mutations can occur gradually in somatic cells over a number of years, leading to the development of a “sporadic” case of cancer. Alternatively, it is possible to inherit dysfunctional genes, leading to the development of a familial form of a particular cancer. A model of cancer development in the colon is shown in Fig. 3.3. This scheme is based largely on observation and genetic analyses of tissues obtained from patient colon at various stages of disease.
Figure 3.3 Colorectal cancer as a model of multistage carcinogenesis – the adenoma–carcinoma sequence. Sequential acquisition of mutations in various genes associated with initiation and progression of cancer are shown for the chromosome instability (CIN) model and the microsatellite instability (MSI) model. However, this does not mean that all colonic cancers arise in this sequence – activation of oncogenes and inactivation of tumor suppressor pathways are the key factors (alternative gene mutations could achieve the same net effect).
Genetic alterations can be placed into two large categories. The first category comprises changes that alter only one or a few nucleotides along a DNA strand, termed point mutations, the second comprises various major rearrangements in genes or entire or parts of chromosomes (this is discussed in more detail in Chapter 10).
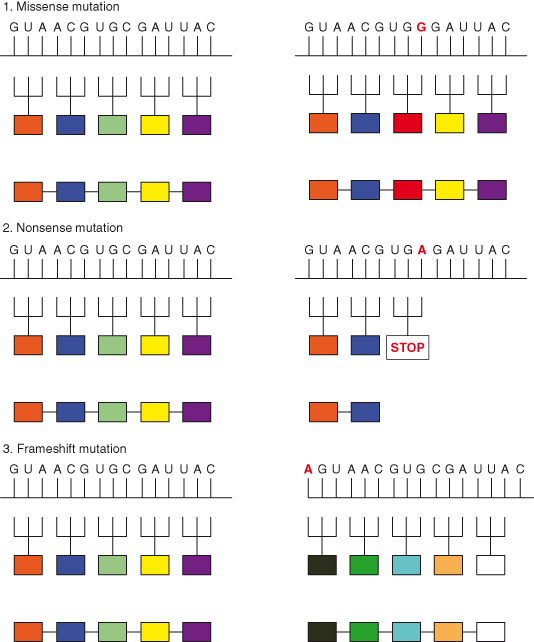
The structure of proteins is encoded in the nucleotide sequence of DNA. A particular sequence of nucleotides gives rise to a particular sequence of amino acids, and that in turn determines the way that that protein will function. Many changes in the nucleotide sequence will alter the amino acid sequence of the protein, and perhaps will change its function. There are several different forms of mutations, with different causes. Mutations can be classified under various headings and are briefly described below and in Fig. 3.4.
Figure 3.4 DNA point mutations. For simplicity, the small part of a mRNA transcribed from the normal or mutated genes is shown and the DNA (gene) itself is not shown. The DNA can be assumed to have a complementary sequence to the small section of mRNA shown. The mutated base is shown in red.
Single-Base Substitutions
A single base is substituted by another, termed a point mutation. If a purine (adenosine or guanine) or a pyrimidine (cytosine or thymine) is replaced by the other member of the same class, the substitution is called a transition, whereas if a purine is replaced by a pyrimidine or vice versa, the substitution is called a transversion. This can happen if DNA polymerase mismatches two bases during replication. There are “proofreading” functions that correct most such errors, but about one in a million is not detected and becomes incorporated permanently in the DNA. Some chemicals, particularly those that alter base structure, greatly increase the chance of DNA polymerase inserting an inappropriate base on the opposite strand, and thus a mutation can result.
Missense Mutations
If the new nucleotide alters the codon, thus producing an altered amino acid in the protein product, this is a missense mutation. One of the three nucleotides making up the codon is replaced and this results in an altered amino acid in the protein product after translation.
Nonsense Mutations
If the new nucleotide changes a codon that specified an amino acid to one of the STOP codons (TAA, TAG, or TGA), resulting in the premature arrest of RNA translation and a truncated protein product, this is a nonsense mutation.
Insertions and Deletions
Extra base pairs may be added (insertions) or removed (deletions) from a gene. These may have major consequences if only one or two bases are involved, as translation of the gene is “frameshifted.” Altering the reading frame one nucleotide to the right or left will result in multiple alterations in the amino acid sequence as multiple codons will now be altered and the mRNA is translated in new groups of three nucleotides. Frameshifts may also create new STOP codons and thus generate nonsense mutations. Alterations in three nucleotides or multiples of three may be less serious because they preserve the reading frame.
Silent Mutations
Most amino acids are encoded by several different codons. Thus TCT, TCG, TCA, and TCC all code for the amino acid serine. Any mutation altering the base at position 3 will have no effect on the resultant protein. Such mutations are silent and detected only by gene sequencing.
Splice-Site Mutations
Intronic sequences are removed during the processing of pre-mRNA to mature mRNA under the influence of various proteins acting at the splice site. If a mutation alters one of these signals, then the intron is not removed and remains as part of the final RNA molecule. The translation of its sequence alters the sequence of the protein product.
More Substantive DNA Mutations
Chromosomal Translocations
In some cases, diseases result not from changes in individual genes but in changes in the number or arrangement of chromosomes. Inherited abnormalities in chromosomes are unusual because they are generally incompatible with normal in utero development. One notable exception is Down syndrome, where individuals have three copies of chromosome 21 instead of the usual two. However, it is in cancers that one primarily finds altered numbers of chromosomes (see also Chapter 10). Some examples are shown in Box 3.3.
Gene Amplification
Sometimes, instead of a single copy of a region of a chromosome, many copies are produced, resulting in the production of multiple copies of genes on that region of the chromosome. In extreme cases, these copies may form their own small pseudo-chromosomes called “double-minute chromosomes.” This is often observed in cancer cells and can result in deregulated expression of oncogenes. Examples of this include amplification of c-MYC in several tumors and amplification of NEU in breast cancers. Gene amplification in the MDR gene encoding the MDR (multiple drug resistance) protein contributes to drug resistance in cancer. The MDR protein is a membrane pump capable of eliminating chemotherapeutic agents from the cancer cell, rendering them ineffective.
Inversions
DNA fragments are sometimes released from a chromosome and then reinserted in the opposite orientation. These inversions may either activate an oncogene or deactivate a tumor suppressor gene.
Duplications/Deletions
Through replication errors, a gene or group of genes may be copied more than once within a chromosome. Duplications are a doubling of a section of the genome. During meiosis, crossing over between sister chromatids that are out of alignment can produce one chromatid with a duplicated gene and the other having two genes with deletions. However, unlike gene amplification, genes are not replicated outside the chromosome and only single copies are produced. Similarly, genes may become lost. Gene duplication has occurred repeatedly during the evolution of eukaryotes – genome analysis reveals many genes with similar sequences in a single organism. If two or more such paralogous genes are still similar in sequence and function, their existence provides redundancy. This may be a major reason why knocking out certain genes in yeast or mice may have little or no effect on phenotype.
Aneuploidy
Entire chromosomes may be lost or replicated during cell division if the replicated chromosomes fail to separate into the daughter cells accurately.
Inherited Susceptibility to Cancer
It was the first time it had ever occurred to me that this detestable cant of false humility might have originated out of the Heep family. I had seen the harvest, but had never thought of the seed.
Charles Dickens, David Copperfield
In inherited diseases, the disease-causing mutation is present at birth and all cells in the body have a mutation. Although most cancers are believed to arise from mutations occurring in single somatic cells in the adult, several inherited cancer syndromes have been described. In these cases, all somatic cells carry a mutation that does not cause cancer on its own, as additional somatic mutations are also needed. Importantly, inherited mutations usually predispose to the development of specific cancers, suggesting that certain cell types in certain tissues exhibit different sensitivity to cancer-susceptibility genes. Because the DNA is already damaged, cells have a headstart when accumulating subsequent mutations, and individuals with an inherited mutation consequently have a much higher risk of getting cancer compared to the general population and develop cancer at a younger age. An example of this for breast cancer is shown in Fig. 3.5.
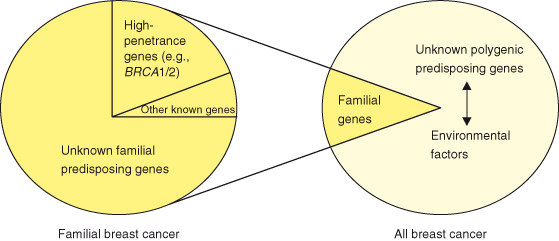
Figure 3.5 Breast cancer susceptibility. Familial breast cancer accounts for around 5–10% of all breast cancers. However, known genes, such as BRCA1 and BRCA2 account for only 20% of the familial risk, so that most genetic factors contributing to breast cancer are unknown. These unknown genetic variants (probably at multiple different loci) likely interact with environmental factors in the pathoetiology of around 80% or more of breast cancers.
Adapted from Balmain, A., Gray, J., and Ponder, B. (2003) The genetics and genomics of cancer. Nat Genet 33(Suppl): 238–44.
By the sixteenth century, both physicians and families were clearly aware that certain overtly visible phenotypic features tended to cluster in families. Even though it would be some time before the scientific basis of this phenomenon would be unraveled, it was known that diseases such as cancer ran in families. Cancer syndromes, such as von Recklinghausen’s neurofibromatosis, which has very evident cutaneous manifestations, were described, although both the honorific and attribution to germline inheritance of one defective copy of the NF1 tumor suppressor gene came much later (the other allele being eventually lost in some somatic cells).
Over the last few decades remarkable progress has been made, leading to the identification of numerous genes that contribute to germline inheritance of cancer susceptibility. Some examples are listed here.
- A hereditary predisposition to breast and ovarian cancer is frequently the result of germline mutations in genes involved in the DNA damage response or in repair of DNA double-strand breaks. Mutations in the BRCA1 and BRCA2 genes are the most important in clinical practice, but recent studies are resulting in identification of further genes, such as two RAD51 paralog–encoding genes, RAD51C, and for ovarian cancer, RAD51D. The presence of such mutations might also identify a subgroup of patients likely to respond favorably to PARP inhibitors (see Chapter 10).
- Lynch syndrome, caused by mutations in the mismatch repair genes MSH2, MLH1, and MSH, is associated with colorectal cancer.
- Familial adenomatous polyposis (FAP), caused by germline mutations in APC, is associated with colonic polyps and colorectal cancer.
- The Li–Fraumeni syndrome, caused by inherited inactivation of one copy of p53 in the germline (occasionally CHEK2), increases the risk of cancers of the breast and brain as well as leukemias and sarcomas.
- Retinoblastoma is caused by inactivation of a copy of the retinoblastoma RB gene.
Other conditions associated with germline-inherited susceptibilities, include:
- Cowden syndrome, due to loss of PTEN, is associated with benign and malignant tumours in the breast, gastrointestinal tract, uterus, ovaries, and thyroid;
- Wilms tumor, associated with mutations in WT1;
- Werner syndrome, caused by mutations in WRN;
- hereditary diffuse gastric cancer, caused by mutations in CDH1;
- Von Hippel–Lindau syndrome, caused by mutations in VHL;
- variants of xeroderma pigmentosum (see Chapter 10) caused by a variety of mutations in DNA excision repair genes, such as ERCC2–5;
- susceptibility to types of leukemia conferred by a variety of genes encoding proteins of the Fanconi anemia complementation group;
- Peutz–Jeghers syndrome, characterized by intestinal hamartomas and increased epithelial cancers, caused by germline mutation in LKB1 (serine/threonine kinase 11);
- familial melanoma caused by germline mutations in CDKN2A, both INK4a (p16) and ARF (p14), or CDK4.
A more comprehensive list is shown in Table 3.1.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


