Fig. 14.1
Schematic of Selecta’s SVP nanoparticle platform SVP nanoparticles are created from polymers [poly(lactic-co-glycolic acid) (PLGA) and polyethylene glycol (PEG) (as PLGA-PEG)] that are present in multiple FDA-approved products. Targeted nanoparticles carry a dense array of surface B-cell antigen (e.g., nicotine) for high avidity B-cell receptor triggering. A TLR agonist adjuvant and a universal T-cell help peptide (TCHP) are encapsulated in the particle core and designed to be released in the endosomal compartment of antigen-presenting cells (APC) where they can trigger TLR receptors and be loaded onto MHC class II complexes, respectively. The physicochemical properties (e.g., size and surface properties) of the nanoparticles target them to desired cell types (APCs) and anatomic locations (lymph nodes)
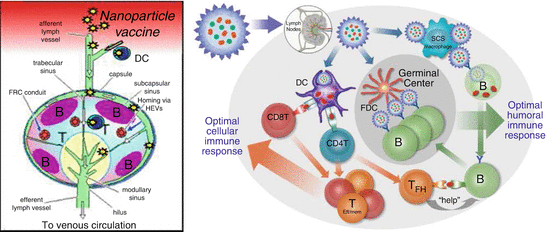
Fig. 14.2
Stylized schematic of local draining lymph node and the interaction among immune cells. (a) Pathogens or particles move via the afferent lymphatics to local draining LNs where they enter through the subcapsular sinus. (b) Once particles have moved into the lymph node, they are captured by dendritic cells (DCs) and subcapsular sinus (SCS) macrophages. DCs internalize and digest the nanoparticles and present payload-derived antigenic peptides in MHC classes I and II to CD8+ and CD4+ T cells, respectively. Activated T cells differentiate into effector/memory (T Eff/Mem ) cells that mediate cellular immune responses. Activated CD4+ T cells also give rise to follicular T helper cells (T FH ). They provide help to B cells that were initially stimulated by surface antigen on nanoparticles captured by SCS macrophages. Following the initial antigen encounter, B cells acquire and process T-cell antigen for restimulation of TFH cells. The action of TFH cells allows the development of a germinal center reaction during which B cells interact with nanoparticles on follicular dendritic cells (FDCs). This induces them to proliferate, generate high-affinity IgG antibodies, and differentiate into memory and plasma cells. All the key players must be provided with the proper information and instruction to generate the highest possible antibody titer and durability
(a)
A particulate form
(b)
Nano-scale size for unimpeded access to lymph vessels and travel to LNs
(c)
Pathogen-associated molecular patterns that serve as adjuvant by stimulating APC-expressed receptors, such as TLRs
(d)
Densely arrayed surface molecules that are capable of cross-linking B-cell receptors (BCRs) on antigen-specific B cells
(e)
Incorporation of protein sequences that can be processed and presented in diverse major histocompatibility (MHC) class II complexes for recognition by CD4+ T cells
14.5.2.3 Hapten Selection
Low molecular weight haptens, such as nicotine (Mw = 162), are not immunogenic and must be attached to high molecular weight carriers or displayed on a nanoparticulate in order to induce an immune response. Earlier studies have shown that nicotine coupled to protein carriers is immunogenic (Pentel et al. 2000; Cerny 2005). In our initial developmental studies, we demonstrated that presentation of nicotine as an antigen covalently attached to SVP results in a potent anti-nicotine antibody response. The self-assembly nature of the SVP platform effectively and efficiently displays the nicotine on the surface of the SVP in a controllable and tunable manner. Nicotine is conjugated to the hydrophilic PEG end of the amphiphilic polymer PLGA–PEG, creating PLGA–PEG–Nic. The SVP nanoparticles are produced by an oil-in-water emulsion process, in which nano-sized oil droplets containing the PLGA–PEG–Nic and other agents are formed. The PLGA–PEG–Nic molecules move by Brownian motion in the oil droplet, and when it nears the oil water interface, the hydrophilic PEG–Nic end of the polymer orients into the water phase where it now stays. In this way the PLGA–PEG–Nic is oriented to display the PEG–Nic end on the surface of the particle, allowing for presentation to B cells. In addition, by varying the amount of the PLGA–PEG–Nic in the formulation, the density of nicotine on the surface of the particle can be varied and optimized to achieve a maximal immune response to nicotine.
14.5.2.4 Adjuvant Selection and Development
Nanoparticles targeted to dendritic cells (DC) may result in more efficient B- and T-cell priming, especially if containing TLR agonists which act via pattern recognition receptors (PRRs), thus serving as signals for DC maturation (Andersen and Ohlfest 2012). Upon being activated through various PRRs, professional APC starts to actively migrate into secondary lymphoid organs, capture antigens, process, and present them, thus driving T- and B-cell activation. In particular, CD4+ cells ensure optimal responses by many other lymphocyte classes, including antibody-producing B cells (Swain et al. 2012).
Several lines of evidence point that effective antigen exposure on the surface of VLP or NP leads to an augmented immunogenicity of a poorly immunogenic epitope (De Filette et al. 2008; Denis et al. 2008; Hashemi et al. 2012). Additionally, utilization of a modular nanoparticle platform makes it possible to accommodate a variety of antigenic iterations for their rapid testing and development. Nanoparticles are typically prepared from organic building blocks and polymers, such as poly-lactide-co-glycolide (PLGA), or inorganic and metallic elements. Biodegradable NP made from PLGA or polylactic acid (PLA) has especially attracted much attention as vehicle for delivery of drugs and vaccines in vivo (Mahapatro and Singh 2011). It has been demonstrated that nanoparticles may target distinct APC with smaller (20–200 nm) particles being able to drain freely to LN-resident DC and macrophages (Manolova et al. 2008; Reddy et al. 2007).
Over the last decades, many strong novel adjuvants have been described, but only one TLR agonist-based composition, AS04, has become in an approved product in the United States (Alving et al. 2012). The main reason for this is the inability to formulate these adjuvants in such a way that their systemic effects are minimized. Since APCs preferentially take up micro- and nanoparticles, using particle-encapsulated adjuvants may solve this safety versus efficacy issue. In fact, many TLR agonists are known to have potent adjuvant properties, but their clinical use has been limited by systemic adverse reactions or insufficient breadth of immune response (e.g., TLR4 agonists are more biased toward Th2 response). It is apparent that efficient encapsulation of adjuvants within the SVPs may enable the safe use of potent TLR agonists.
TLRs are widely expressed on dendritic cells (DC) and other professional APCs such as macrophages and B cells. While some TLRs are expressed on the cell surface and act as sensors for extracellular PAMPs (e.g., lipopolysaccharides), a subset of TLR molecules (TLR3, TLR7, TLR8, and TLR9) are expressed on endosomal membranes and bind nucleic acid-derived molecules, such as single-stranded RNA of viral origin for TLR7 and TLR8 (Hemmi et al. 2002; Jurk et al. 2002; Edwards 2003; Diebold 2004, 2008; Heil et al. 2004) and bacterial unmethylated DNA oligonucleotides (ODNs) containing CpG motifs (CpG ODNs) for TLR9 (Krieg et al. 1995; Sato 1996; Roman et al. 1997; Sparwasser et al. 1997; Hemmi et al. 2000). TLR ligands of natural and synthetic origin are potent inducers of innate immune responses and have been shown to effectively stimulate the transition from an innate immune response to an adaptive immune response. As such, TLR agonists have been evaluated as potential adjuvants in a variety of applications (Wang and Singh 2011).
To date, only one PRR ligand, 3-O-desacyl-4’-monophosphoryl lipid A (MPL), a TLR4 agonist, has been included as an adjuvant in an FDA- or EMA-licensed vaccine. MPL adsorbed onto alum is utilized in the HPV vaccine Cervarix, licensed in the United States and Europe (Harper 2009), and the hepatitis B vaccine Fendrix, licensed in Europe (Johnson 2008). Imiquimod, a topically administered TLR7 agonist, has been approved for treatment of genital warts, actinic keratosis, and basal cell carcinoma (Basith et al. 2011). Other TLR agonists, such as poly (I:C) (TLR3), imidazoquinolines other than imiquimod (TLR7, TLR8, or TLR7/TLR8), and CpG ODNs (TLR9), have failed thus far to enter clinical practice as parenteral adjuvants despite a multitude of promising data obtained in preclinical and clinical studies (Wang et al. 2005; Krieg 2006; Wille-Reece et al. 2006; Karan et al. 2007; Krieg 2007). One of the main reasons for this failure is the delicate balance between the induction of augmented immunogenicity by TLR agonists and safety concerns, which are often related to the generation of systemic inflammatory responses (Hemmi et al. 2002; Vasilakos et al. 2000; Doxsee et al. 2003; Pockros 2007).
Several groups have utilized micro- and nanocarriers, such as viruslike particles, liposomes, and PLGA particles, to encapsulate adjuvants (Bachmann and Jennings 2010; Malyala et al. 2009; Kasturi et al. 2011). Encapsulation of adjuvants reduces systemic exposure of adjuvant and enhances uptake by APCs. Nano-sized viruses and particles distribute rapidly to the local draining lymph node where they are taken up by subcapsular macrophages and dendritic cells (Manolova et al. 2008; Bachmann and Jennings 2010; Junt 2007). Antigens can also be delivered in particles to target efficient uptake by APCs (Bachmann and Jennings 2010; Krieg 2007; Singh et al. 2006; Wagner 2009). Recent studies show that co-encapsulation of both antigen and adjuvant in nanoparticulate carriers has synergistic effects in augmenting immunity by targeting antigen and adjuvant to the endosomal compartment of APCs (Krieg 2007; Kasturi et al. 2011; Wagner 2009).
We evaluated the immune responses induced by synthetic vaccine particles (SVP) carrying covalently bound or entrapped TLR agonist co-delivered with encapsulated antigen (either in the same or in separate nanoparticle preparations). We hypothesized that such an approach may provide a two-pronged benefit by enabling a focused delivery of antigen and adjuvant and hence enhancing immunogenicity while preventing systemic exposure of the TLR agonist, which can result in excessive systemic cytokine release. We were able to demonstrate that the SVPs deliver their payloads directly to APCs in lymph nodes, as evidenced by the local production of cytokines in draining lymph nodes and the lack of systemic inflammatory cytokines that are typically associated with adverse reactions.
Nanoparticle Encapsulation of TLR Agonists Changes the Pattern of Inflammatory Cytokine Induction In Vitro
TLR7/TLR8 (resiquimod also known as R848) and TLR9 (CpG ODN 1826; mouse-specific B-type CpG ODN) agonists were encapsulated in synthetic polymer nanoparticles and tested for their ability to induce cytokines in vitro. R848 was chemically conjugated to PLGA and used for SVP formulation as PLGA-R848, and CpG ODN was passively entrapped into SVP as described by Ilyinskii et al. (Ilyinskii et al. 2014). Natural oligonucleotide sequences contain a phosphodiester (PO) backbone, which is susceptible to rapid hydrolytic cleavage by nucleases in vivo. Nuclease-resistant CpG sequences with a phosphorothioate (PS) backbone have been shown to have superior activity to PO-CpG in vivo. Both PS and PO forms of the immunostimulatory CpG ODN 1826 sequence (PS-CpG 1826 and PO-CpG 1826) were evaluated. SVP-encapsulated R848 induced lower levels of TNF-α from splenocytes and especially from murine macrophages than identical amounts of free R848 (Fig. 14.3a, b). Similar profiles were seen when PS-CpG 1826 and PO-CpG 1826 sequences were tested in free or SVP-encapsulated form. Not surprisingly, PO-CpG 1826 was a less potent inducer of TNF-α production than PS-CpG 1826, with its SVP-encapsulated form being nearly inactive, even in the more sensitive J774 cells (Fig. 14.3c, d). IL-6 production in vitro followed the same pattern as TNF-α (data not shown). However, a static in vitro system does not capture potential differences in biodistribution and pharmacokinetics of free adjuvant versus nanoparticle-encapsulated adjuvant that is expected in vivo.
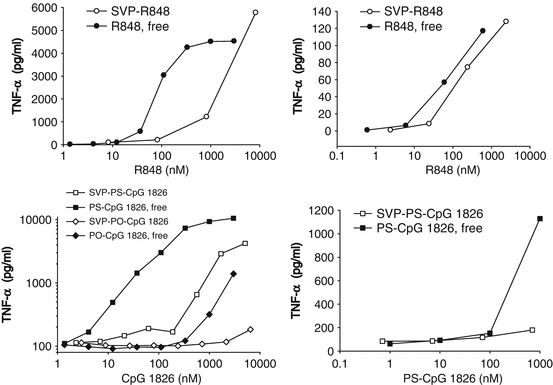
Fig. 14.3
Adjuvant encapsulation in nanoparticles (SVP) results in suppressed TNF-α induction in murine cells in vitro. SVP containing TLR7/TLR8 agonist R848 (a, b) or TLR9 agonists PO-CpG 1826 (c) or PS-CpG1826 (d) were added to J774 cells (a, c) or fresh mouse splenocyte cultures (b, d) in parallel with free adjuvants in triplicates. The amount of TNF-α in culture supernatants was measured 6 h (splenocytes) or 16 h (J774) after incubation. Representative results out of two separate experiments with groups of three mice are shown
Rapid Infiltration of Draining Lymph Nodes by Innate and Adaptive Immune Cells Upon Injection of Nanoparticle-Encapsulated R848
As reported by Ilyinskii et al. (Ilyinskii et al. 2014), R848-bearing nanoparticles induced a profound increase in cellularity within the draining lymph nodes at 4 days after a single inoculation (Fig. 14.4). Further analysis of cellularity within the draining lymph nodes after s.c. injection showed that LN infiltration starts as early as 1 day after inoculation, reaches a peak at 7–8 days, and is maintained for at least 3 weeks (Tables 14.1 and 14.2). The increase in lymph node cellularity was even more rapid and pronounced in mice that were previously immunized with SVP (tenfold increase in the popliteal LN cell count at 3 days after inoculation, Table 14.2). No significant cell infiltration of the draining lymph node was seen if SVP lacking R848 were used either alone or admixed with free R848 (Table 14.1).
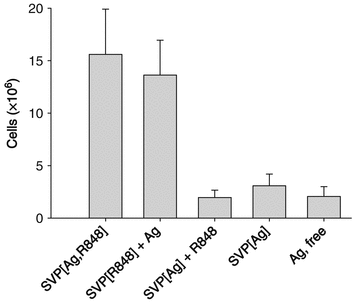
Fig. 14.4
Nanoparticle encapsulation of TLR7/TLR8 agonist (adjuvant) results in higher local immune infiltration than utilization of free adjuvant. Total draining lymph node (LN) cell count is presented at 4 days after s.c. injection. The total dose of antigen (Ag) was the same across all treatment groups, and the total dose of R848 was the same for all groups receiving R848
Table 14.1
Local lymphadenopathy induction by administration of SVP-encapsulated but not free TLR7/TLR8 agonist
Day | SVP-R848 | SVP (no adjuvant) | SVP (no adjuvant) + R848 |
|---|---|---|---|
0 | 1.00 | 1.00 | 1.00 |
1 | 1.50 | 1.36 | 1.08 |
4 | 3.19 | 1.53 | 1.03 |
7 | 8.06 | 1.53 | 0.84 |
Table 14.2
Local lymphadenopathy after administration of SVP-encapsulated TLR7/TLR8 agonist and nicotine antigen in naive vs. pre-immunized mice
SVP-R848, total cells in LN | ||
|---|---|---|
Day | Naive mice | Immune mice |
0 | 1.00 | 1.00 |
3 | 4.33 | 10.14 |
8 | 8.04 | 9.86 |
14 | 5.36 | 6.93 |
21 | 4.14 | 6.60 |
A detailed analysis of intranodal cell populations after injection of SVP containing both encapsulated R848 and nicotine antigen (Nic) showed a rapid increase in the number of innate immune cells, such as granulocytes and myeloid DC, in the draining LN, with their numbers increasing threefold within 24 h after a single injection (Table 14.3). There was also an early elevation in macrophage cell numbers in the draining lymph node, while increases in other APC subtypes (plasmacytoid DC and B cells) were observed at a slightly later time point. Interestingly, among the populations analyzed, only effector cells of the adaptive immune response (T and B cells) showed a continued expansion from day 4 to day 7 (Table 14.3). Presence of nicotine antigen within the SVP had no influence on LN cellularity.
Table 14.3
Local lymphadenopathy and immune cell population expansion after administration of SVP-encapsulated TLR7/TLR8 agonist irrespective of nicotine presence
Day | Total cells | CD11c+ B220– (mDC) | CD11c+B220+ (pDC) | B220+ CD11c– (B cells) | F4/80+ GR1– (Mø) | F4/80– GR1+ (Gran) | CD3+ (T cells) | CD3–CD49b+ (NK) | CD3+ CD49b+ (NKT) |
|---|---|---|---|---|---|---|---|---|---|
0 | 1.00 | 1.00 | 1.00 | 1.00 | 1.000 | 1.00 | 1.00 | 1.00 | 1.00 |
1 | 1.50 | 3.15 | 1.36 | 0.98 | 1.97 | 3.52 | 1.52 | 1.03 | 0.62 |
4 | 3.19 | 6.50 | 8.14 | 3.56 | 1.16 | 14.20 | 2.54 | 9.64 | 6.72 |
7 | 8.06 | 7.00 | 7.77 | 11.09 | 2.51 | 18.43 | 4.39 | 8.57 | 8.25 |
Nanoparticle Encapsulation of TLR7/TLR8 Agonist Induces Strong Local Cytokine Production
Strong local immune activation by nanoparticle-encapsulated R848 was further manifested by cytokine production in the draining LN milieu (Figs. 14.5 and 14.6). At 4 h after subcutaneous injection, high levels of IFN-γ, RANTES, IL-12(p40), and IL-1β were secreted by LNs from animals injected with SVP[Ag,R848] (encapsulated R848 and antigen), while the production of these cytokines by LNs from mice injected with free R848 was close to the background level (Fig. 14.5). In particular, the amount of IFN-γ secreted by LNs from mice injected with SVP[Ag,R848] at 4 h was more than 100-fold higher than that from the LNs of mice injected with SVP[Ag] and an equal amount of free R848 (Fig. 14.5a), while RANTES was elevated more than 27-fold (Fig. 14.5b). Production of all of these cytokines in the LN was maintained for at least 72 h after injection of SVP[Ag,R848], with levels of IL-12(p40) and IL-1β remaining nearly stable (Fig. 14.5c, d) and levels of IFN-γ and RANTES, while decreasing, remaining 4- to 20-fold higher than the background. In contrast, inoculation of free R848 led to only a modest increase of local cytokine production at 4 h, which returned to background levels by 24 h after administration (Ilyinskii et al. 2014). Levels of IP-10 and MCP-1 in LNs from SVP[Ag,R848]-injected animals were also elevated in a similar fashion (data not shown).
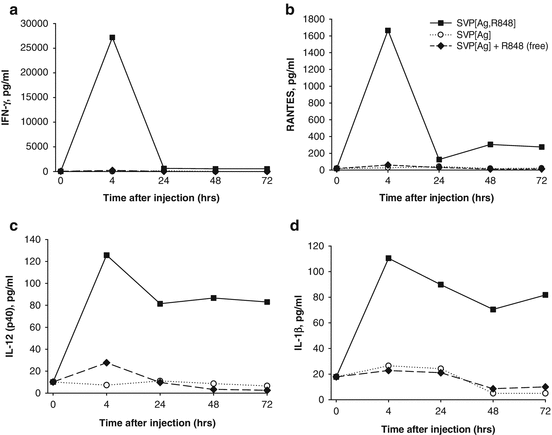
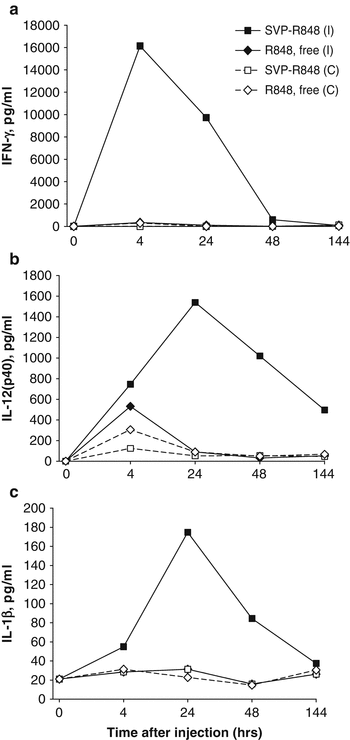
Fig. 14.5
Local cytokines are induced by SVP-encapsulated, but not free, TLR7/TLR8 agonist R848. Mice were injected s.c. in both hind limbs with either SVP-OVA-R848, SVP-OVA, or SVP-OVA admixed with free R848. At the times indicated, popliteal LNs were taken and incubated in vitro overnight. Culture supernatants were collected and analyzed for the presence of specific cytokines by ELISA. (a) IFN-γ, (b) RANTES, (c) IL-12(p40), (d) IL-1β. Average of four LNs per time point is shown
Fig. 14.6
Focused local cytokine induction after a single-site injection with SVP encapsulated, but not free, TLR7/TLR8 agonist R848. Mice were injected s.c. in a single hind limb with either SVP-R848 or free R848. At the times indicated, popliteal LNs from the injection side (ipsilateral, I) and from the opposite side (contralateral, C) were collected and incubated in vitro overnight. Culture supernatants were analyzed for the presence of specific cytokines by ELISA. (a) IFN-γ, (b) IL-12(p40), (c) IL-1β. Average of two mice per time point is shown
The striking difference in local cytokine production after administration of nanoparticle-encapsulated versus free R848 (Fig. 14.5) was also evident by comparing cytokine production in the ipsilateral draining lymph node versus the contralateral lymph node after injection in a single hind limb (Figs. 14.6a, b). The sustained expression of IFN-γ, IL-12(p40), and IL-1β was seen in the ipsilateral LN at 4–48 h after injection of SVP-R848, but not in the contralateral lymph node. In contrast, free R848 induced a modest elevation of IL-12(p40) and IFN-γ in both the ipsilateral and contralateral lymph nodes (Fig. 14.6b). The level of IFN-γ observed in the ipsilateral lymph node following injection of free R848 was 50-fold lower than that induced by SVP-R848 (Fig. 14.6a). No induction of IL-1β by free R848 was seen (Fig. 14.6c) (Ilyinskii et al. 2014).
Nanoparticle Encapsulation of TLR7/TLR8 Agonist R848 Attenuates the Production of Systemic Inflammatory Cytokines
While nanoparticle encapsulation of R848 enhanced immunogenicity and local induction of immune cytokines, the production of systemic inflammatory cytokines by SVP-R848 was markedly suppressed compared to that observed with free R848 after either subcutaneous or intranasal inoculation (Figs. 14.7 and 14.8, respectively). In particular, 4 h after subcutaneous inoculation, serum concentrations of early inflammatory cytokines TNF-α and IL-6 were 50–200 times higher if free R848 was used (Fig. 14.7a, b). Serum cytokine levels were similar in animals inoculated with SVP[Ag] (antigen-containing SVP) with or without encapsulated R848. Similar differences were observed with systemic production of RANTES (Fig. 14.7c). SVP[Ag,R848] induced modest levels of IP-10, IL-12(p40), and MCP-1, which were approximately five to ten times lower than that observed after injection of SVP[Ag] admixed with free R848 (Figs. 14.7d–f) (Ilyinskii et al. 2014).
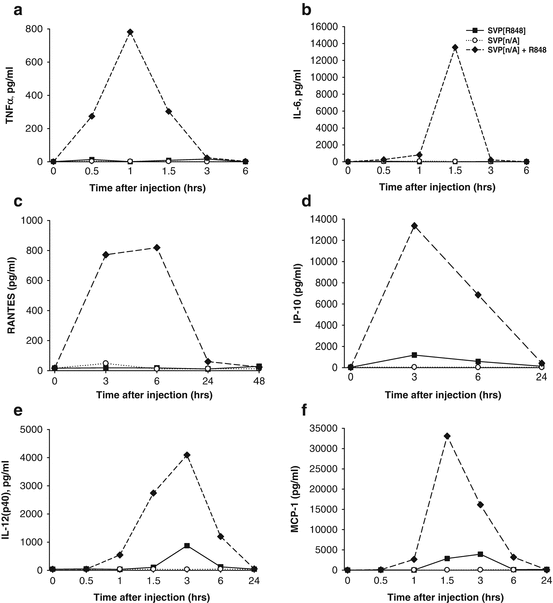
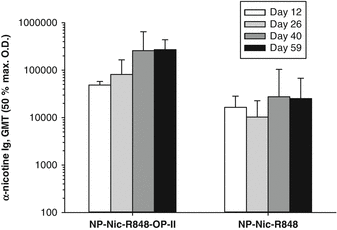
Fig. 14.7
Strong systemic cytokine induction after injection of free, but not SVP-encapsulated TLR7/TLR8 agonist R848. Mice were injected s.c. with either SVP[R848], SVP without adjuvant (SVP[n/A]), or SVP[n/A] admixed with free R848 and bled at times indicated. The same amount of free or SVP-encapsulated R848 was used for each group (6.8 μg). Serum samples were collected at the times indicated and analyzed for individual cytokines by ELISA. Average cytokine concentration is shown (three samples per group per each time point). (a) TNF-α, (b) IL-6, (c) RANTES, (d) IP-10, (e) IL-12(p40), (f) MCP-1
Fig. 14.8
T helper peptide OP-II provides for stronger immunogenicity upon prime and boost with SVP. Mice were immunized subcutaneously with either NP[Nic,R848] or NP[Nic,R848,OP-II] on days 0, 14, and 28. Serum was collected at days 12, 26, 40, and 59, and antibody titers to nicotine were measured by ELISA. NP[Nic,R848,OP-II] induced a significantly higher anti-nicotine antibody response than NP[Nic,R848] without OP-II (Mann–Whitney rank sum test, p < 0.005). Bars on graph represent anti-nicotine antibody geometric mean titers (GMT) with standard deviations (n = 5 mice/group)
14.5.2.5 T Helper Peptide Development
A T-cell helper antigen is required for effective generation of anti-nicotine antibodies by SVP since in the absence of so-called T-cell help, IgG affinity maturation and Ig class switching do not occur. Briefly, T lymphocytes can be separated into two subpopulations based on their expression of the cell surface markers CD4 and CD8. The CD4+ subset is primarily responsible for providing “help” to stimulate and activate other immune cells through direct cell–cell interactions and/or the secretion of cytokines. Collaboration with B cells leads to an antibody isotype switch and enhanced antibody production. The T-cell receptors of CD4+ T cells recognize peptide antigens presented by MHC class II molecules on APCs, such as DCs. MHC class II molecules generally present peptides derived from exogenous antigens. Exogenous antigens are captured by DCs and trafficked into the endo-/lysosomal compartment for processing. The resulting peptides are loaded onto MHC class II molecules and presented to MHC class II-restricted CD4+ helper T cells in the draining lymph nodes.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


