Myeloid Sarcoma
Definition
A tumor mass of immature myeloid cells involving an extramedullary site or bone (1). The neoplasm can precede, occur concurrently, or arise subsequent to the diagnosis of acute myeloid leukemia. Myeloid sarcoma also can arise in patients with myelodysplastic syndromes, chronic myeloproliferative diseases, and myelodysplastic/myeloproliferative diseases.
Synonyms
Chloroma; myeloblastoma; extramedullary myeloid cell tumor; granulocytic sarcoma; monocytic sarcoma.
Epidemiology and Pathogenesis
The epidemiology of patients with myeloid sarcoma is essentially that of patients with acute myeloid leukemia (AML), either de novo, or in patients with myelodysplastic syndromes (MDS), chronic myeloproliferative diseases (CMPD), or myelodysplastic/myeloproliferative diseases (MDS/MPD). Acute myeloid leukemia is primarily a disease of adults and is the most frequent form of leukemia in adults in the Western world (2,3). The median age at time of diagnosis in various studies varies from 60 to 65 years of age (1,3). The incidence of AML in persons less than 65 years is 1.8 per 100,000 persons. In persons older than 65 years, this incidence has been reported to be as high as 17 per 100,000 persons (3). The high incidence in the elderly is associated with the high incidence of associated myelodysplasia (1,3). There is also a small peak in AML incidence in very young children, but the frequency of AML in this age group is far outnumbered by acute lymphoblastic leukemia.
Geographical variation occurs in the incidence of AML, with the highest incidences in the United States, Australia, and western Europe (3). In the United States, it is estimated that 11,930 new cases of AML occurred in 2006. There is a slight male predominance. In the 2006 estimate of new cases of AML, 6,350 men and 5,580 women developed AML for a male-to-female ratio of 1.14:1 (3). New cases of AML are slightly more common in whites than African-Americans. If untreated, AML is a fatal disease, but with new therapies a subset of patients are now cured. The AML mortality rate is substantially higher in older than in younger patients, and is slightly higher for males than females. In the United States, it is currently estimated that approximately 7,800 adults die of AML per year (3,4).
The etiology of AML is poorly understood. Ionizing radiation, either as a result of inappropriate exposure (e.g., atomic bombs in Hiroshima and Nagasaki) or used therapeutically, is known to increase the risk of AML (1,3). Chemotherapy also increases the risk of AML, with cytotoxic agents and topoisomerase II inhibitors inducing AML via different mechanisms. Exposure to certain chemicals, such as benzene and pesticides and herbicides, is a risk factor. Cigarette smoking increases the risk of developing AML twofold. Inherited diseases are also associated with an increased risk of AML; these include Fanconi anemia, Down syndrome, Klinefelter syndrome, Li-Fraumeni syndrome, ataxia-telangiectasia, neurofibromatosis, and others (3). Despite knowledge of these risk factors, most cases of AML occur de novo, without known exposure to risk factors.
Myeloid sarcoma is uncommon in patients with AML, MDS, CMPD, or MDS/MPD. In an older study of 478 patients with myeloid leukemia, including AML and chronic myelogenous leukemia, myeloid sarcoma occurred in approximately 3% of patients (5). In more recent studies, a similar frequency of myeloid sarcoma is reported. Breccia and colleagues reported their experience in Italy; myeloid sarcoma occurred in 2% of patients with AML (6). In both of these studies, many patients had myeloid sarcoma with concurrent AML (5,6). At M.D. Anderson Cancer Center, 1,520 patients with AML and 402 patients with high-risk MDS seen between 1990 until 2002 were reviewed. Twenty-one patients in this group presented with myeloid sarcoma without a history or concurrent evidence of AML, MDS, CMPD, or MDS/MPD. This group represented 1.1% of all patients (7). Despite therapy appropriate for AML, approximately 25% of these patients went on to develop AML, with a median interval of 5 months (7).
The pathogenetic mechanisms involved in a myeloid neoplasm presenting as myeloid sarcoma, rather than AML, are unknown. Furthermore, the explanation for a myeloid sarcoma arising in a patient with AML is also not understood. What is known is that patients with certain types of AML are more prone to develop myeloid sarcoma. These include patients with the core binding factor leukemias, AML with t(8;21)(q22;q22) and acute myelomonocytic leukemia with inv(16)(p13q22), as well as patients with acute monoblastic/monocytic leukemia (1,8,9). Chronic myelomonocytic leukemia, now considered a MDS/MPD in the World Health Organization (WHO) classification, also is associated with a relatively high frequency of myeloid sarcoma (10). Others have suggested that expression of CD56 may correlate with risk of myeloid sarcoma (11). In one study, expression of the T-cell–associated antigens CD2 and CD7 by myeloblasts was suggested as being associated with a greater chance of developing myeloid sarcoma (12), but this was not confirmed in subsequent larger studies (13).
Clinical Syndrome
The age and sex ratio of patients with myeloid sarcoma mirrors that of patients with AML, MDS, CMPD, or MDS/MPD. There is a wide age range, from childhood to adults, but adults are more common, and males and females are equally affected. One potential difference may be median age. As stated earlier, the median age for patients with AML and other myeloid diseases is over 60 years. In contrast, Byrd and colleagues (8) reviewed the literature and identified 154 cases of myeloid sarcoma with a median age of 36 years. However, selection bias and other factors may also be involved that could explain this discrepancy.
Myeloid sarcoma occurs in patients with AML in three clinical settings: (a) Most often, myeloid sarcoma is associated with concurrent evidence of AML involving the blood and bone marrow; (b) myeloid sarcoma can arise in patients with a history of
AML as a sign of relapse (in this setting, myeloid sarcoma can be isolated or associated with blood and bone marrow disease); and (c) least frequently, myeloid sarcoma can arise in patients without a history or concurrent evidence of AML. If untreated, many of these patients go on to develop AML within less than 1 year, but therapy appropriate for AML can be curative. In patients with MDS, CMPD, or MDS/MPD, the development of myeloid sarcoma indicates that the disease has transformed to a blast phase (blast crisis), and is often accompanied by the presence of AML in blood and bone marrow (14). Prognosis is generally poor in this patient group.
AML as a sign of relapse (in this setting, myeloid sarcoma can be isolated or associated with blood and bone marrow disease); and (c) least frequently, myeloid sarcoma can arise in patients without a history or concurrent evidence of AML. If untreated, many of these patients go on to develop AML within less than 1 year, but therapy appropriate for AML can be curative. In patients with MDS, CMPD, or MDS/MPD, the development of myeloid sarcoma indicates that the disease has transformed to a blast phase (blast crisis), and is often accompanied by the presence of AML in blood and bone marrow (14). Prognosis is generally poor in this patient group.
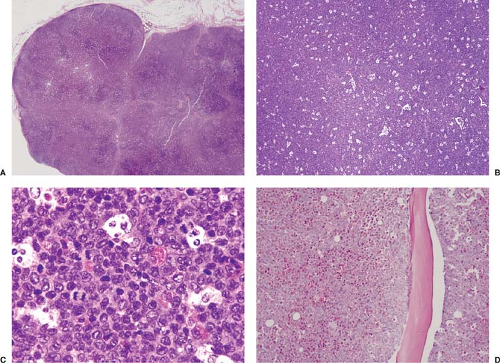 Figure 76.1. Myeloid (granulocytic) sarcoma involving lymph node. A: At low-power magnification, the lymph node is subtotally replaced. B: At this magnification, a starry-sky pattern can be easily appreciated. C: At high-power magnification, the neoplastic cells are immature but show evidence of differentiation, as shown by the presence of eosinophilic myelocytes and metamyelocytes. D: Bone marrow biopsy specimen showing extensive replacement of the medullary space by acute myeloid leukemia. The neoplasm in the bone marrow was shown to carry the inv(16)(p13q22) by conventional cytogenetics. The diagnoses of myeloid sarcoma in lymph node and bone marrow involvement by acute myeloid leukemia were made almost simultaneously. A–D, Hematoxylin-eosin stain. |
Myeloid sarcoma can occur in virtually any anatomic site with symptoms and signs related to the exact location and size of the mass, or to the presence of concurrent AML. The most common sites of myeloid sarcoma, as stated in the WHO classification, are the subperiosteal bone structures of the skull, paranasal sinuses, bones (sternum, ribs, vertebrae, and pelvis), lymph nodes (Fig. 76.1), and skin (1). However, others have reported these and other sites being involved with different frequencies. For example, Tsimberidou and colleagues reported that skin and lymph nodes are the most common sites of myeloid sarcoma (7). There are also ample case reports and small case series in the literature describing myeloid sarcoma involving the central nervous system, spinal cord, head and neck region, breast, heart, thymus, liver, spleen, pancreas, gastrointestinal tract (Fig. 76.2), endocrine glands, genitourinary system (Fig. 76.3), gynecologic tract, and other sites (15,16,17,18,19,20,21,22,23,24,25,26,27). For extramedullary sites of involvement, some correlation also exists between location and/or patient age with specific type of myeloid neoplasm. For example, in children, the orbit is commonly involved (17), and chronic myelomonocytic leukemia appears to have a strong tropism for skin involvement, often at the terminal stages of the disease (10).
Gross Pathology
Myeloid sarcomas were initially designated as chloroma because many tumors, particularly those with granulocytic differentiation, exhibit a characteristic green color. This color is the result of abundant myeloperoxidase (initially called verdoperoxide), a green peroxidative enzyme present in the cytoplasmic granules of the neoplastic cells. Typically, the green color fades on exposure to air within 2 to 3 hours. However,
the color may be restored by applying hydrogen peroxide or sodium metabisulfite and preserved by storing the specimen in glycerol (28). Using ultraviolet light, myeloid sarcomas exhibit bright red fluorescence because of the presence of abundant protoporphyrins.
the color may be restored by applying hydrogen peroxide or sodium metabisulfite and preserved by storing the specimen in glycerol (28). Using ultraviolet light, myeloid sarcomas exhibit bright red fluorescence because of the presence of abundant protoporphyrins.
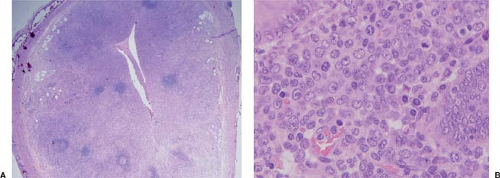 Figure 76.2. Myeloid (granulocytic) sarcoma involving appendix in a child initially thought clinically to have appendicitis. A: The neoplasm extensively replaces the mucosa of the appendix. B: At high-power magnification, most of the neoplastic cells are immature but a rare eosinophilic myelocyte is present in this field. Acute myeloid leukemia developed within 1 week of the appendectomy. Hematoxylin-eosin stains. |
Histopathology
Myeloid sarcoma involving any extramedullary site forms a mass that invades and displaces normal tissue structures (28). In patients with AML involving blood and bone marrow, extramedullary sites may be involved by the neoplasm as small perivascular aggregates of cells. These aggregates, although evidence of AML, do not meet the criteria for myeloid sarcoma.
In lymph nodes, myeloid sarcoma replaces the architecture with sheets of myeloblasts (Fig. 76.1) (29,30). In lymph nodes with partial involvement, myeloid sarcoma may infiltrate the sinuses, paracortex, and medulla, sparing residual germinal centers (Fig. 76.1 A–D). Cytologically, myeloid sarcomas can reflect the full spectrum observed in AMLs. The most common form of myeloid sarcoma, also known as granulocytic sarcoma, is composed of blasts with medium to large nuclei with multilobated, round, reniform, or folded contours; small to inconspicuous nucleoli; and a high nucleus-to-cytoplasm ratio (28,30). Mitotic figures are usually numerous. Foci of necrosis can occur. Auer rods cannot be seen in routinely prepared hematoxylin-eosin tissue sections.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


