CHAPTER 27 Myelodysplastic/myeloproliferative neoplasms
Introduction
Myelodysplastic/myeloproliferative neoplasms (MDS/MPN) are clonal hematopoietic malignancies with hybrid myelodysplastic and myeloproliferative features. In these disorders, cytopenias and dysplasia may coexist with elevated white blood cell counts, thrombocytosis and/or organomegaly making it difficult to assign individual cases into a myelodysplastic or myeloproliferative category. Even though disorders with mixed myelodysplastic and myeloproliferative features have been recognized for many years, the precise diagnosis and assignment to a biologically defined category has been challenging. Acknowledging the unique characteristics of these diseases, in 2001 the World Health Organization classification of hematopoietic and lymphoid malignancies created a separate group of MDS/MPN.1 Chronic myelomonocytic leukemia (CMML) is perhaps the most well known example of a MDS/MPN. Depending on WBC count, CMML may resemble more closely a myelodysplastic syndrome (MDS) or a myeloproliferative neoplasm (MPN). The disease is clinically heterogeneous and lacks a precise biological connotation. Similar to CMML, other categories of MDS/MPNs are also a genetically heterogeneous group of diseases with variable patient outcomes. They are frequently positioned on a continuum between MDS and MPN, and thus are challenging to diagnose using traditional morphology-based approaches. Integration of morphologic features with laboratory data, clinical presentation, immunophenotype and cytogenetic/molecular tests is required to categorize MDS/MPNs according to current WHO 2008 classification.2 It is critical to diagnose patients at the time of original presentation, i.e. before bone marrow (BM) and peripheral blood (PB) features are modified by therapy or progression of the disease. If a patient has been previously diagnosed elsewhere, a complete review of the original diagnostic material is mandatory. ‘Re-classification’ based on the review of post-treatment and/or follow-up BM sample is strongly discouraged.
Three discrete MDS/MPN entities can be reliably classified using the above described integrated multiparametric approach: chronic myelomonocytic leukemia (CMML), atypical chronic myeloid leukemia (aCML), BCR-ABL1 negative and juvenile myelomonocytic leukemia (JMML). Occasionally, a myeloid neoplasm with myelodysplastic and myeloproliferative features cannot be assigned to any of these ‘classical’ categories. Such cases should be diagnosed as MDS/MPN, unclassifiable.2 This group includes also the provisional entity known as refractory anemia with ring sideroblasts associated with marked thrombocytosis (RARS-T). Additionally, there are rare disorders, which may qualify for inclusion in the MDS/MPN group in the future. One such disease is known as myeloid neoplasm with an isolated isochromosome 17q. This is a rare entity with a rapidly progressive clinical course that may occasionally morphologically resemble CMML.
A detailed discussion of the MDS/MPNs is presented below. The main diagnostic features of these neoplasms are summarized in Table 27.1. Some cytogenetic and genetic features are also addressed in Chapter 21.
Chronic myelomonocytic leukemia
Definition and diagnostic criteria
CMML is a hematopoietic malignancy with hybrid myeloproliferative and myelodysplastic features. The diagnostic criteria include: 1) persistent unexplained monocytosis (>1 × 109/l with >10% monocytes on the differential count); 2) absence of defining genetic features such as BCR-ABL1 fusion or rearrangements of PDGFRA and PDGFRB; 3) less than 20% blasts; and 4) dysplasia in one or more myeloid lineages. In the absence of definitive dysplastic features, the diagnosis of CMML can be rendered if unexplained monocytosis persists longer than 3 months or if cytogenetic/molecular studies demonstrate the presence of acquired clonal cytogenetic or molecular genetic abnormality.2
Epidemiology, clinical and laboratory features
CMML is a rare disease with estimated incidence of 3 cases per 100 000 people.3 The disease is more prevalent in men with a predominance of 1.5–3 : 1 and has a median age of presentation between 65 and 75 years.4,5 The disease is more common in Western countries as compared to Asian population.6 Patients can present with elevated WBC or with leukopenia. Signs and symptoms of BM failure are common and include fatigue, susceptibility to infections and bleeding. Weight loss, night sweats, fever, organomegaly, tissue infiltration (e.g. skin lesions) and serous effusions are also seen. Depending on the WBC (threshold 13 × 109/l), CMML has been previously divided into myeloproliferative or myelodysplastic categories.7 Splenomegaly and increased LDH are more common in the myeloproliferative subtype of CMML.8,9 Other clinical and laboratory features, and prognosis, including overall survival and incidence of transformation into acute leukemia, are similar for both groups.8–10 Whether the myelodysplastic and myeloproliferative CMML should be considered different phases of the disease or represent distinct entities is at present unclear. The separation is not required by the WHO 2008 classification.
Morphology and immunophenotypic features
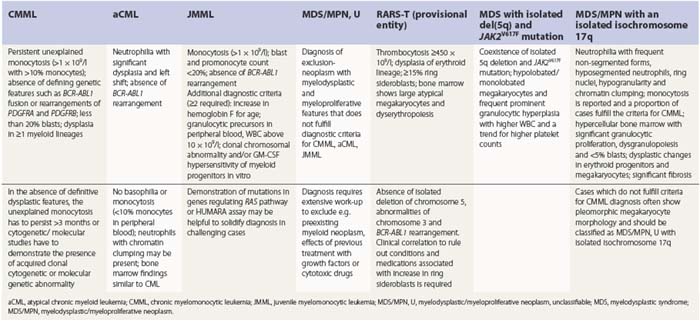
PB shows significant monocytosis (>1 × 109/l with >10% monocytes on the differential count), variable WBC (neutrophilia is seen in about 50% of the cases), anemia and thrombocytopenia. The majority of monocytes are mature, and they frequently display abnormal nuclear and cytoplasmic features. These include abnormal nuclear lobulation or chromatin pattern, prominent granulation or increased basophilia of cytoplasm (Fig. 27.1). Morphological definition of ‘abnormal monocytes’ and promonocytes has been a matter of debate and is thoroughly described in the WHO 2008 classification.2 In most cases, peripheral blood blasts, including monoblasts and promonocytes constitute less than 5% of the differential count. Neutrophilia may be present with dysgranulopoiesis including pseudo-Pelger–Huët cells and hypogranulation. Circulating neutrophilic precursors (promyelocytes and myelocytes) usually constitute less than 10% of white blood cells. In cases with significant eosinophilia (≥1.5 × 109/l), molecular studies excluding the rearrangements in PDGFRA and PDGFRB genes are necessary to further consider the diagnosis of CMML. Basophilia is uncommon. Giant platelets can be seen. Red blood cells are more often normochromic and normocytic but macrocytosis can also be seen.
Even though peripheral blood monocytosis is the single most important diagnostic finding, the final diagnosis of CMML should never be rendered without BM evaluation. The population of promonocytes, which are morphologically more differentiated than blasts in the bone marrow is not infrequently present in PB of patients with acute myelomonocytic and monocytic leukemia. Also, increase in monocytes largely confined to the BM without absolute monocytosis in PB can be seen in rare cases of ‘marrow predominant’ CMML. These diagnoses can be missed if a BM examination is not performed. Conversely, significant monocytosis can also be seen in some cases of BCR-ABL1+ chronic myelogenous leukemia (CML), which should always be excluded (see Chapter 24).
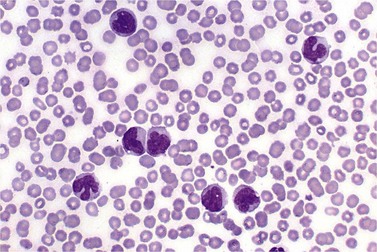
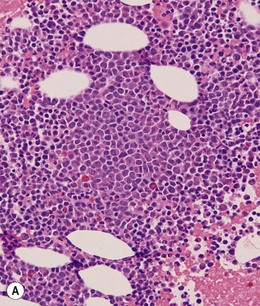
BM is significantly hypercellular in a majority of CMML cases, a finding easily appreciated in a predominantly elderly population, in which this disease occurs (Fig. 27.2A). Rarely, BM may be normo- or hypocellular.11 A prominent myeloid proliferation is usually seen (Fig. 27.2B). This includes both an increased number of neutrophilic forms as well as monocytes, which can be difficult to appreciate in the absence of non-specific esterase cytochemistry. In a significant number of cases, the granulopoiesis may be left shifted and show significant dysplasia including hypogranulation and nuclear abnormalities.7 Due to difficulties in differentiating monocytes from immature granulocytic cells (e.g. myelocytes and metamyelocytes), the quantification of the monocytic component may be challenging, which causes a significant variability between observers. The most significant practical issue is the differentiation of abnormal monocytes from monoblasts and promonocytes. The early stages of monocytic differentiation in the bone marrow of CMML patient are shown in Fig. 27.2C.
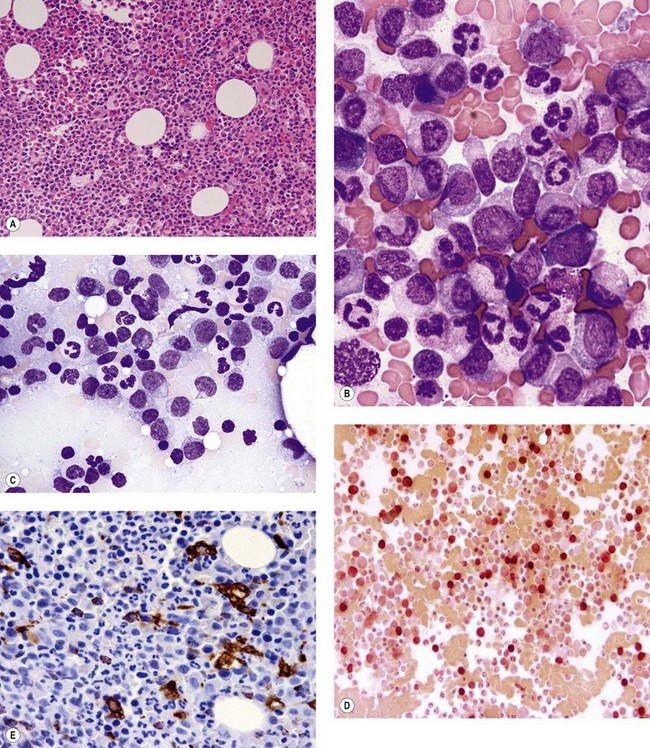
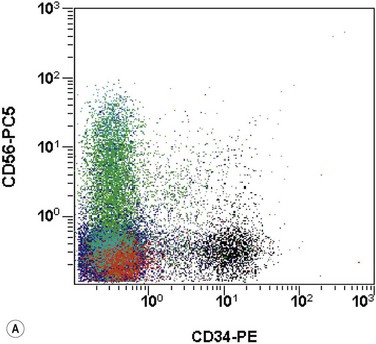
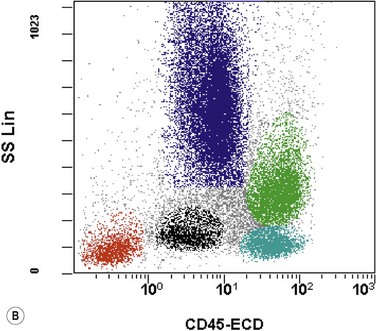
The application of nonspecific esterase and immunophenotyping by flow cytometry or immunohistochemistry are helpful in quantifying the monocytic component. Nonspecific esterase staining (alpha-naphthyl butyrate or alpha-naphthyl acetate esterase) is an effective method of demonstrating monocytic differentiation. The stain is useful to confirm the increased number of esterase positive cells in cases of CMML. Median values of 10–20% esterase positive cells are found in the majority of CMML patients12,13 (Fig. 27.2D). Multiparametric flow cytometry has the added benefits of separating various stages of monocytic differentiation and demonstrating an aberrant antigen expression. The former can be accomplished by profiling the expression of the different epitopes of CD14 antigen in combination with CD64 and CD33, which reveals the presence of promonocytes and more mature monocytic forms.14 Aberrant expression of myeloid antigens, both loss and altered antigen density, and expression of non-myeloid markers, are frequently seen in monocytes and in the granulocytic population. Monocytes show altered density of HLA-DR, CD13 and CD64, and may show co-expression of CD2 and CD56 (Fig. 27.3A). In comparison to reactive monocytes, the aberrant expression of CD56 on CMML monocytes was much higher.15,16 The decrease in right-angle light scatter is not infrequent and reflects the hypogranulation of granulocytic series (Fig. 27.3B). Increase in the number of CD34 and/or CD117 positive blasts may indicate progression of the disease and alert to the impending transformation.
With the exception of demonstrating an increased number of blasts and blast clusters using immunohistochemical stain for CD34, the value of immunohistochemistry in CMML has been limited. Common myeloid/monocyte/macrophage immunohistochemical markers such as lysozyme and CD68 (KP1) applied to BM trephine biopsies (BMTB), are generally not reliable in quantifying monocytic component.12 CD68R and CD163 have been shown to be more restricted to monocytes and macrophages. However, these markers usually stain significantly lower numbers of monocytic cells as compared to naphthyl butyrate esterase12,17 (Fig. 27.2E). Some investigators reported that staining pattern of CD68R (fine vs coarse granules) in conjunction with cell morphology can discriminate between monocyte precursors and mature monocytes.18 The success of this approach may, however, be dependent on the processing of BMTB and staining methodology used, and needs further validation and standardization in a multi-institutional setting. New antibodies applicable to paraffin embedded material have recently been reported to aid in enumeration of the monocytic component in BMTB. A combined use of CD14 and CD16 allowed discriminating between monocyte population and dysplastic granulocytes in this setting.17
Numerous studies confirmed that the number of blasts and promonocytes is the most reliable predictor of patient prognosis and of the risk of transformation to acute leukemia. Recognizing the importance of the increase in blasts, CMML is divided into two groups: CMML-1 with less than 5% blasts in PB and less than 10% in BM; and CMML-2 with 5–19% of blasts in PB and 10–19% in BM. The prognostic value of this classification has been previously confirmed.19
Erythropoiesis is usually normal to decreased and may show dysplastic changes.20 These include megaloblastoid change with asynchrony in maturation of nucleus and cytoplasm, and nuclear abnormalities such as nuclear fragmentation, irregularity of nuclear outline and multinucleation. Increased megakaryopoiesis is not uncommon; however, it is usually less pronounced than that seen in cases of BCR-ABL1+ CML.12 Dysmegakaryopoiesis is seen in the majority of CMML cases.21 Dysplastic megakaryocytes display abnormal nuclear lobation including mononuclear or hypolobated forms, or separated nuclear lobes. Micromegakaryocytes can also be seen. Approximately 20% of cases show significant reticulin fibrosis.13
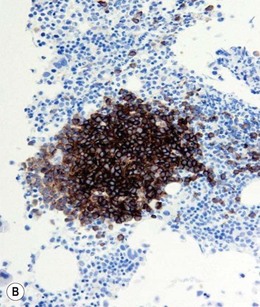
Approximately 20% of CMML cases show nodular proliferations of plasmacytoid dendritic cells12 (Fig. 27.4). The lineage and origin of these cells have long been debated and is reflected by changing nomenclature starting from the designation of ‘T-associated plasma cells’ and ‘plasmacytoid T-cells’ to ‘plasmacytoid monocytes’. Currently, plasmacytoid dendritic cells are believed to be derived from myeloid progenitors. Thus, it is perhaps not surprising that focal proliferations of these cells are seen in association with myeloid neoplasms with monocytic differentiation, most commonly in CMML. These nodules are considered clonal and in select cases have been shown to be clonally related to the underlying myeloid neoplasm.22,23 Plasmacytoid dendritic cells are intermediate in size with round to oval nucleus, with or without indentation, finely dispersed chromatin and moderately abundant cytoplasm with distinct cytoplasmic border. Their identification is facilitated by immunohistochemical stains including CD123, CD68, CD68R, CD4 and CD14. Plasmacytoid dendritic cells are also positive for granzyme B and occasionally for CD56, CD2 and CD5. They are negative for other lymphoid markers, and for CD13, CD11c, myeloperoxidase and CD34.
Prognosis
The survival of patients with CMML is extremely variable, ranging from 1 to 100 months.2,4,12,24 Considerable efforts have been made in the past to design a predictor of clinical outcome. All scoring systems shown to be useful in predicting the clinical outcome (such as modified Bournemouth score, Dusseldorf score, MD Anderson prognostic score and Spanish CMML score) used a combination of BM blast count with PB counts to predict survival.5,24 In multivariate analysis, PB and BM blast percentage proved to be the strongest predictor of survival and of transformation into acute myeloid leukemia (AML).4,13,19,25,26 PB and BM blast counts are the keystones to distinguish between CMML-1 and CMML-2. These two subcategories were shown to be highly predictive of overall survival and correlated well with transformation into AML. Specifically, at 5-year follow-up, 63% of patients with a BM blast count of 10% or greater developed AML as compared to 18% of patients with blast count below 10%.19 Previously discussed myelodysplastic and myeloproliferative subtypes of CMML were not significant for predicting patient outcome.19
As our knowledge on the pathogenesis of myeloid neoplasms expands, new prognostic factors emerge. TET2 gene mutations have been recently reported to occur in 50% of CMML patients (see Chapter 21) and have been associated with poor survival in the CMML-1 subgroup.27 RUNX1 mutations occur in up to 40% of CMML cases and abnormalities of C-terminal portion of this gene are associated with progression to AML.28
Special considerations
Chronic myelomonocytic leukemia developing in a course of myelodysplastic syndrome
The evolution of previously diagnosed MDS to CMML is well documented.29–32 The pathogenesis of this phenomenon is not well understood with only rare studies testing select genes implicated in myeloid neoplasms.32 The interval between the initial MDS diagnosis and the evolution to CMML ranged between 2 and 59 months.
Approximately 20% of MDS patients show relative peripheral blood monocytosis (more than 10% monocytes but with absolute monocyte count less than 1 × 109/l) at the time of their initial diagnosis. Over time, a significant proportion of these patients may develop CMML. Conversely, up to 24% of patients with CMML have a documented preceding MDS phase.13,32 Both groups, whether originally presenting with or without relative monocytosis, initially show less than 1 × 109/l monocytes in PB. Rigolin et al. reported the distinct clinicopathological features of MDS cases with relative monocytosis (>10% monocytes).29 In comparison to MDS without monocytosis, these patients showed higher marrow cellularity, higher incidence of chromosomal abnormalities, and a more aggressive disease with high incidence of progression to CMML and transformation into acute myelomonocytic or monocytic leukemia.29 When CMML evolving from the MDS phase was compared to myelodysplastic type of de novo CMML, the former showed a superior survival.31,32
Therapy-related CMML
The majority of CMML cases represent de novo disease arising in patients without a pre-existing hematologic or non-hematopoietic neoplasm. Occasionally, CMML may develop after treatment with chemotherapeutic agents and/or radiotherapy for malignancies or non-neoplastic diseases.33–35 Even though the numbers of reported cases are low, therapy-related CMML seems to show morphologic and immunophenotypic features similar to those of de novo CMML. However, a proportion of these cases show the rearrangement of MLL gene, a finding which is usually seen only in acute leukemia. Regardless of morphologic features, current WHO classification recommends to classify all therapy-related cases under the umbrella of therapy-related myeloid neoplasms. Therefore, all therapy-related CMML cases should be included in this category. Thus, the detailed knowledge of patient’s previous medical history is critical while diagnosing CMML.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


