Musculoskeletal Disorders
INTRODUCTION
A complex system of bones, muscles, ligaments, tendons, and other connective tissue, the musculoskeletal system gives the body its form and shape. It also protects vital organs, makes movement possible, stores calcium and other minerals, and provides sites for hematopoiesis. A fibrous layer called the periosteum covers all bones, except at joints, where they’re covered by articular cartilage.
The human skeleton contains 206 bones, which are composed of inorganic salts, such as calcium and phosphate, embedded in a framework of collagen fibers. Bones are classified by shape as long, short, flat, or irregular.
LONG BONES
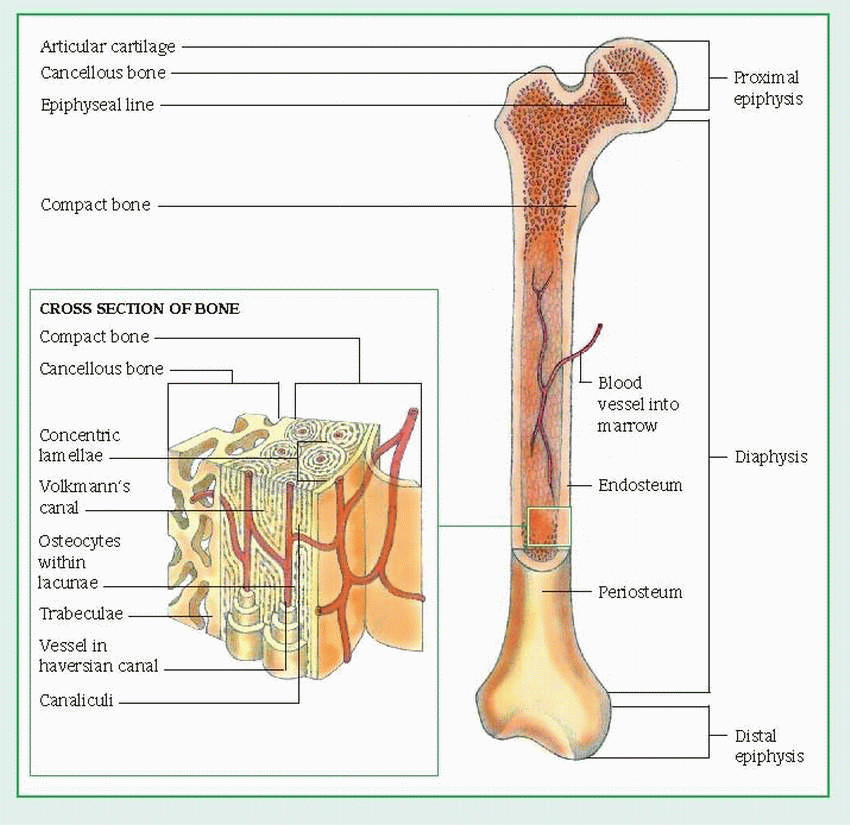
Long bones, which are found in the limbs, include the humerus, radius, and ulna of the arm; the femur, tibia, and fibula of the leg; and the phalanges, metacarpals, and metatarsals in the hands and feet. These bones have a long shaft, or diaphysis, and widened, bulbous ends, called epiphyses. A long bone is made up mainly of compact bone, which surrounds the medullary cavity (also called the yellow marrow), a storage site for fat. The lining of the medullary cavity (the endosteum) is a thin layer of connective tissue. The outer layer is the periosteum. (See Long-bone structure, page 296.)
In children and young adults, lengthwise growth occurs at the epiphyseal cartilage between the diaphysis and epiphysis. In adults, in whom bone growth is complete, this cartilage is ossified and forms the epiphyseal line. The epiphysis also has a surface layer made up of compact bone, but its center is made of spongy or cancellous bone. Cancellous bone contains open spaces between thin threads of bone, called trabeculae, which are arranged in various directions to correspond with the lines of maximum stress or pressure. This configuration gives the bone added structural strength.
Unlike cancellous bone, adult compact bone consists of numerous orderly networks of interconnecting canals that run parallel to the bone’s long axis. Each of these networks, called a haversian system, consists of a central haversian canal surrounded by layers (lamellae) of bone. Between adjacent lamellae are small openings (lacunae), which contain bone cells (osteocytes). All lacunae are joined by an interconnecting network of tiny canals (canaliculi), each of which contains one or more capillaries and provides a route for movement of tissue fluids. The haversian system carries blood to the bone through blood vessels that enter the system through channels called Volkmann’s canals.
SHORT, FLAT, OR IRREGULAR BONES
Short bones include the tarsal and carpal bones; flat bones, the frontal and parietal bones of the cranium, ribs, sternum, scapulae, ilium, and pubis; and irregular bones, the bones of the spine (vertebrae, sacrum, and coccyx) and certain bones of the skull (sphenoid, ethmoid, and mandible).
Short, flat, and irregular bones have an outer layer of compact bone and an inner portion of spongy bone. In the sternum and certain areas in the flat bones of the skull, the spongy bone contains red marrow.
JOINTS
The tissues connecting two bones make up a joint, which permits motion between the bones and provides stability. Joints, like bones, have varying forms.
Fibrous joints (synarthroses) have only minute motion and provide stability when tight union is necessary, as in the seams, called sutures, that join the cranial bones.
Cartilaginous joints (amphiarthroses) have limited motion, as between vertebrae and symphysis pubis.
Synovial joints (diarthroses) are the most common and have the greatest degree of movement. Such joints include the elbows, shoulders, and knees. Synovial joints have special characteristics: the articulating surfaces of each bone have a smooth hyaline covering (articular cartilage), which is resilient to pressure; their opposing surfaces are congruous and glide smoothly on each other without touching each other; a fibrous (articular) capsule holds them together. Beneath the capsule and lining the joint cavity, the synovial membrane secretes the clear, viscous synovial fluid. This fluid lubricates the two opposing surfaces during motion and also nourishes the articular cartilage. Surrounding a synovial joint are ligaments, muscles, and tendons, which strengthen and stabilize the joint but allow free movement.
In some synovial joints, the synovial membrane forms two additional structures—bursae and tendon sheaths—which reduce friction that normally accompanies movement. Bursae are small, cushionlike sacs lined with synovial membranes and filled with synovial fluid; most are located between tendons and bones. Tendon sheaths wrap around the tendon and cushion it as it crosses the joint.
The synovial joints permit angular and circular movements. Angular movements include flexion (decrease in joint angle), extension (increase in the joint angle), and hyperextension (increase in the angle of extension beyond the usual arc). Joints of the knees, elbows, and phalanges permit such movement. Other angular movements are abduction (movement away from the body’s midline) and adduction (movement toward the body’s midline).
Circular movements include rotation (motion around a central axis), as in the ball-and-socket joints of the hips and shoulders; pronation (wrist motion to place palmar surface of the hand down, with the thumb toward the body); supination (begging position, with palm up). Other kinds of movement are inversion (movement facing inward), eversion (movement facing outward), protraction (as in forward motion of the mandible), and retraction (returning protracted part into place).
MUSCLES
Muscle tissues’ most specialized feature— contractility— makes movement of bones and joints possible. Muscles also pump blood through the body, move food through the intestines, and make breathing possible. Muscular activity produces heat, so it’s an important component in temperature regulation. Muscles maintain body positions, such as sitting and
standing. Muscle mass accounts for about 40% of the body weight of a person of average size.
standing. Muscle mass accounts for about 40% of the body weight of a person of average size.
Muscles are classified in many ways. Skeletal muscles are attached to bone, visceral muscles permit function of internal organs, and cardiac muscles make up the heart wall. Also, muscles may be striated or nonstriated (smooth), depending on their cellular configuration.
Muscles classified according to activity are called voluntary or involuntary. Voluntary muscles can be controlled at will and are under the influence of the somatic nervous system; these are the skeletal muscles. Involuntary muscles, controlled by the autonomic nervous system, include the cardiac and visceral muscles.
Each skeletal muscle consists of many elongated muscle cells, called muscle fibers, through which run slender threads of protein, called myofibrils. Muscle fibers are held together in bundles by sheaths of fibrous tissue, called fascia. Blood vessels and nerves pass through the fascia to reach the individual muscle fibers.
Skeletal muscles are attached to bone directly or indirectly by fibrous cords called tendons. The least movable end of the muscle attachment is called the point of origin; the most movable end is the point of insertion.
MECHANISM OF CONTRACTION
To stimulate muscle contraction and movement, the brain sends motor impulses through the peripheral motor nerves to motor nerve fibers in the voluntary muscle. These nerve fibers reach membranes of skeletal muscle cells at neuromuscular (myoneural) junctions. When an impulse reaches the myoneural junction, it triggers the following sequence: release of the neurochemical acetylcholine, transient release of calcium from the sarcoplasmic reticulum (a membranous network in the muscle fiber), and muscle contraction. The arriving impulse at the myoneural junction also triggers release of adenosine triphosphate, the energy source for muscle contraction. Muscle relaxation is believed to take place by reversal of the above mechanisms.
MUSCULOSKELETAL ASSESSMENT
Most patients with musculoskeletal disorders are elderly, have concurrent medical conditions, or have experienced trauma. Younger patients tend to experience more benign, self-limited conditions. Generally, they face prolonged immobilization. These factors make thorough assessment essential. Your assessment should include a complete history and a careful physical examination to determine a possible cause of the symptoms.
Interview the patient carefully to obtain a complete medical, social, and personal history. Ask about general activity (does he jog daily, or is he sedentary?), which may be significantly altered by musculoskeletal disease or trauma. Does the patient have any systemic symptoms, such as fever, chills, weight loss, or skin rashes? Obtain information about occupation, diet, sexual activity, and elimination habits, drugs taken, and use of safety devices, and try to assess how the problem will affect body image. Also, ask how he functions at home. Can he perform activities of daily living? Does he have difficulty getting around? Are there stairs where he lives? Where are the bathroom and bedroom? Does he use any prosthetic devices? Ask if other family members can help with his care.
Get an accurate account of the musculoskeletal problem. Ask the patient if it has caused him to change his everyday routine. When did symptoms begin and how did they progress? Has the patient received treatment for this problem? If he’s experienced trauma, find out how he was hurt.
Assess the level of pain. Is the patient in pain at the moment? Ask what makes the discomfort worse or better (movement, position, and so forth). Evaluate past and present responses to treatment. For instance, if the patient has arthritis and uses corticosteroids, ask him about their effectiveness. Does he require more or less medication than before? Did he comply with the prescribed treatment?
The physical examination helps to determine the diagnosis and reveals any existing disabilities. (These baseline data will help when the effects of treatment are evaluated.) Observe the patient’s appearance. Look for localized edema, pigmentation, redness and tenderness at pressure points, and other deformities such as atrophy. Note mobility, strength, and gait. To check range of motion, ask the patient to abduct, adduct, or flex the muscles in question. Obtain height, weight, and vital signs. Check neurovascular status, including motion sensation and circulation. Measure and record discrepancies in muscle circumference or leg length. Compare one side or limb to the other. If a neck injury is suspected, don’t force range of motion.
DIAGNOSTIC TOOLS
X-rays are a useful diagnostic tool to evaluate musculoskeletal diseases. They can help to
identify joint disruption, bone deformities, calcifications, and bone destruction and fractures. X-rays also measure bone density.
Myelography is an invasive procedure used to evaluate abnormalities of the spinal canal and cord. It entails injection of a radiopaque contrast medium into the subarachnoid space of the spine. Serial X-rays visualize the progress of the contrast medium as it passes through the subarachnoid space. Displacement of the medium indicates a space-occupying lesion, such as a herniated disk or a tumor.
Magnetic resonance imaging is useful in evaluating soft-tissue injuries or ligament tears, such as rotator cuff tears or meniscal tears.
Computed tomography scan can be used to identify injuries to bones, soft tissue, ligaments, tendons, and muscles.
Arthroscopy is the visual examination of the interior of a joint with a fiber-optic endoscope.
Other useful tests include bone and muscle biopsies, electromyography, microscopic examination of synovial fluid, and multiple laboratory studies of urine and blood to identify systemic abnormalities.
PATIENT CARE
Each patient with musculoskeletal disease needs an individual care plan formulated early in his hospital stay by the entire clinical team, including the physician, physical therapist, and occupational therapist. Develop this plan with short- and long-term goals, during and after hospitalization.
Caring for the patient with a musculoskeletal disease usually includes at least one of the following: traction, casts, braces, splints, crutches, intermittent range-of-motion devices, prolonged immobilization, physical therapy, occupational therapy, and self-care measures; adequate vitamin D intake, weight loss, dietary modifications, and drugs.
Traction is the manual or mechanical application of a steady pulling force to reduce a fracture, minimize muscle spasms, or immobilize or align a joint.
Skin traction is the indirect application of traction to the skeletal system through skin and soft tissues.
Skeletal traction is the direct application of traction to bones by means of a pin (Steinmann pin) or wire (Kirschner wire) through the affected bone or by calipers or a tonglike device (Gardner-Wells tongs) that grips the bone.
Manual traction, for emergency use, is the direct application of traction to a body part by hand.
During the use of all types of traction:
Explain to the patient how traction works, and advise him about permissible amounts of activity and elevation of the head of the bed. Inform him of the anticipated duration of traction and whether or not the traction is removable. Teach active range-of-motion (ROM) exercises.
Check neurovascular status to prevent nerve damage. Also, make sure the mattress is firm, that the traction ropes aren’t frayed, that they’re on the center track of the pulley, and that traction weights are hanging free. Thoroughly investigate any complaint the patient makes.
Check for signs of infection (odor, local inflammation and drainage, or fever) at pin sites if the patient is in skeletal traction. Also, check with the physician’s or the facility’s procedure regarding pin-site care, such as use of peroxide or povidone-iodine.
Ideally, a cast immobilizes without adding too much weight. It’s snug-fitting but doesn’t constrict and has a smooth inner surface and smooth edges to prevent pressure or skin irritation. Casts require comprehensive patient education.
A plaster cast takes 24 to 48 hours to dry. To prevent indentations, tell the patient not to squeeze the cast with his fingers, not to cover or walk on the cast until it has dried, and not to bump a damp cast on hard surfaces because dents can cause pressure areas. Warn the patient that while the cast is drying, he’ll feel a temporary sensation of heat under the cast.
If fiberglass is used, the cast may feel dry and the patient may be able to bear weight immediately. Advise the patient, however, not to get the cast wet. Although the fiberglass won’t disintegrate as plaster would, the padding will become wet and potentially cause maceration of the skin.
Emphasize the need to keep the cast above heart level for 24 hours after its application to reduce swelling in the limb.
While the cast is drying and after drying is complete, the patient should watch for and immediately report persistent pain in the limb inside or distal to the cast as well as edema, changes in skin color, coldness, or tingling or numbness in this area. If any of these signs occur, tell the patient to position the casted body part above heart level and notify his physician.
The patient should also report drainage through the cast or an odor that may indicate infection. Warn against inserting foreign objects under the cast, getting it wet, pulling out its padding, or scratching inside it. Tell the patient to seek immediate attention for a broken cast.
Instruct the patient to exercise the joints above and below the cast to prevent stiffness and contracture.
Braces, splints, and slings also provide alignment, immobilization, and pain relief for musculoskeletal diseases. Slings and splints are usually used for short-term immobilization. Explain to the patient and his family why these appliances are necessary, and show them the proper way to apply the sling, splint, or brace for optimal benefit. Tell the patient how long the appliance will have to be worn, and advise him of any activity limitations that must be observed. If the patient has a brace, check with his orthotist (orthopedic appliance specialist) about proper care. Encourage the patient to refer additional questions to his physician. Teach proper crutch walking.
COPING WITH IMMOBILITY
Immobilized patients require meticulous care to prevent complications. Without constant care, the bedridden patient becomes susceptible to pressure ulcers, caused by the increased pressure on tissue over bony prominences, and is especially vulnerable to cardiopulmonary complications.
To prevent pressure ulcers, turn the patient every 2 hours and, if possible, position him in a 30-degree side-lying position for short periods. In addition, place a flotation pad or sheepskin pad under bony prominences, or use an alternating-air-current, convoluted foam, or foam mattress. Show the patient how to use a Balkan frame with a trapeze to move about in bed.
Keep the patient’s skin dry and clean.
Keep the sheets wrinkle-free.
Increase fluid intake to minimize risk of renal calculi.
Provide adequate nutrition; a high-protein diet is preferred, if tolerated.
Perform passive ROM exercises on the affected side, as ordered, to prevent contractures, and instruct the patient in active ROM exercises on the unaffected side. Apply footboards or high-topped sneakers to prevent footdrop. Keep the patient’s heels off the bed to prevent heel breakdown. Also, watch for reddened elbows.
Because most bedridden patients involuntarily perform a Valsalva maneuver when using the upper arms and trunk to move, instruct the patient to exhale (instead of holding his breath) as he turns. This will prevent possible cardiac complications that result from increased intrathoracic pressure.
Emphasize the importance of coughing and deep breathing, and teach the patient how to use the incentive spirometer if ordered.
Because constipation is a common problem in bedridden patients, establish a bowel program (fluids, fiber, laxatives, stool softeners), as needed.
REHABILITATION
Restoring the patient to his former state of health isn’t always possible. When it isn’t, help the patient adjust to a modified lifestyle. During hospitalization, promote independence by letting him finish difficult tasks by himself. If necessary, refer the patient to a community facility for continued rehabilitation.
CONGENITAL DISORDERS
Clubfoot
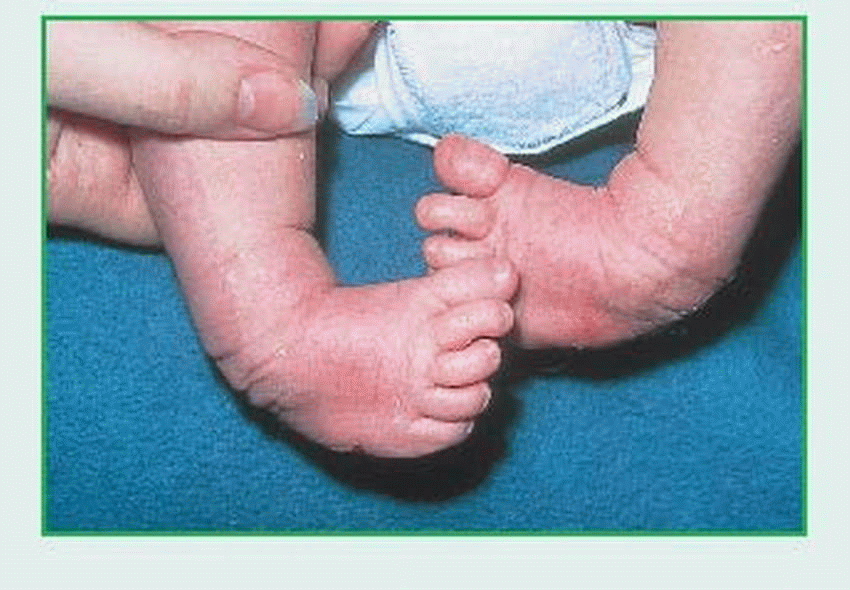
Clubfoot, or talipes, is the most common congenital disorder of the lower limbs. It’s marked primarily by a deformed talus and shortened Achilles tendon, which give the foot a characteristic clublike appearance. In talipes equinovarus, the foot points downward (equinus) and turns inward (varus), whereas the front of the foot curls toward the heel (forefoot adduction).
CAUSES AND INCIDENCE
It is no longer believed that clubfoot is caused by fetal position in utero. Heredity is a definite factor in some cases, although the mechanism of transmission is undetermined. In children without a family history of clubfoot, this anomaly seems linked to arrested development during the 9th and 10th weeks of embryonic life, when the feet are formed. Researchers also suspect muscle abnormalities, leading to variations in length and tendon insertions, as possible causes of clubfoot. Environmental factors play a role. Studies strongly link clubfoot to cigarette smoking during pregnancy, especially if there is a family history of clubfoot.
Clubfoot, which has an incidence of about 1 per 1,000 live births, usually occurs bilaterally and is twice as common in boys. It may be associated with other birth defects, such as myelomeningocele, spina bifida, and arthrogryposis. However, most cases are sporadic occurrences.
COMPLICATION
Retention deformity
SIGNS AND SYMPTOMS
Talipes equinovarus varies greatly in severity. Deformity may be so extreme that the toes
touch the inside of the ankle, or it may be only vaguely apparent. In every case, the talus is deformed, the Achilles tendon shortened, and the calcaneus somewhat shortened and flattened. Depending on the degree of the varus deformity, the calf muscles are shortened and underdeveloped, and soft-tissue contractures form at the site of the deformity. The foot is tight in its deformed position and resists manual efforts to push it into normal position. Clubfoot is painless, except in elderly, arthritic patients. In older children, clubfoot may be secondary to paralysis, poliomyelitis, or cerebral palsy, in which case treatment must include management of the underlying disease.
touch the inside of the ankle, or it may be only vaguely apparent. In every case, the talus is deformed, the Achilles tendon shortened, and the calcaneus somewhat shortened and flattened. Depending on the degree of the varus deformity, the calf muscles are shortened and underdeveloped, and soft-tissue contractures form at the site of the deformity. The foot is tight in its deformed position and resists manual efforts to push it into normal position. Clubfoot is painless, except in elderly, arthritic patients. In older children, clubfoot may be secondary to paralysis, poliomyelitis, or cerebral palsy, in which case treatment must include management of the underlying disease.
DIAGNOSIS
Early diagnosis of clubfoot is usually possible because the deformity is obvious. In subtle deformity, however, true clubfoot must be distinguished from apparent clubfoot (metatarsus varus or pigeon toe). Apparent clubfoot is inversion of the feet, resulting from the peroneal type of progressive muscular atrophy and progressive muscular dystrophy. In true clubfoot, X-rays show superimposition of the talus and the calcaneus and a ladderlike appearance of the metatarsals. (See Recognizing clubfoot.)
TREATMENT
Clubfoot is correctable with prompt treatment, which is performed in three stages: correcting the deformity, maintaining the correction until the foot regains normal muscle balance, and observing the foot closely for several years to prevent the deformity from recurring. In neonates with true clubfoot, corrective treatment should begin at once. An infant’s foot contains large amounts of cartilage; the muscles, ligaments, and tendons are supple. The ideal time to begin treatment is during the first few days and weeks of life, when the foot is most malleable.
Clubfoot deformities are usually corrected in sequential order. Several therapeutic methods have been found effective in correcting clubfoot. In all patients, the first procedure should be simple manipulation and casting, whereby the foot is gently manipulated into a partially corrected position and held in place by a cast for several days or weeks. (The skin should be painted with a nonirritating adhesive liquid beforehand to prevent the cast from slipping.) After the cast is removed, the foot is manipulated into an even better position and casted again. This procedure is repeated as many times as necessary. In some cases, the shape of the cast can be transformed through a series of wedging maneuvers instead of changing the cast each time.
After correction of clubfoot, proper foot alignment should be maintained through exercise, night splints, and orthopedic shoes. With manipulating and casting, correction usually takes about 3 months. The Denis Browne splint, a device that consists of two padded, metal footplates connected by a flat, horizontal bar, is sometimes used as a follow-up measure to help promote bilateral correction and strengthen the foot muscles.
Resistant clubfoot may require surgery. Older children, for example, with recurrent or neglected clubfoot usually need surgery. Tenotomy, tendon transfer, stripping of the plantar fascia, and capsulotomy are some of the surgical procedures that may be used. In severe cases, bone surgery (wedge resections, osteotomy, or astragalectomy) may be appropriate. After surgery, a cast is applied to preserve the correction. Clubfoot severe enough to require surgery is rarely totally correctable; however, surgery can usually ameliorate the deformity.
SPECIAL CONSIDERATIONS
The primary concern is recognition of clubfoot as early as possible, preferably in neonates.
Look for any exaggerated attitudes in an infant’s feet. Make sure you recognize the difference between true clubfoot and apparent clubfoot. Don’t use excessive force in trying to manipulate a clubfoot. The foot with apparent clubfoot moves easily.
Stress to parents the importance of prompt treatment. Make sure they understand that
clubfoot demands immediate therapy and orthopedic supervision until growth is completed.
After casting, elevate the child’s feet with pillows. Check the toes every 1 to 2 hours for temperature, color, sensation, motion, and capillary refill time; watch for edema. Before a child in a clubfoot cast is discharged, teach parents to recognize circulatory impairment.
Insert plastic petals over the top edges of a new cast while it’s still wet to keep urine from soaking and softening the cast. This is done as follows: Cut a plastic sheet into strips long enough to cover the outside of the cast, and tuck them about a finger length beneath the cast edges. Using overlapping strips of tape, tack the corner of each petal to the outside of the cast. When the cast is dry, petal the edges with adhesive tape to keep out plaster crumbs and prevent skin irritation. Perform good skin care under the cast edges every 4 hours, washing and carefully drying the skin. (Don’t rub the skin with alcohol, and don’t use oils or powders, which tend to macerate the skin.)
If the child is old enough to walk, caution parents not to let the foot part of the cast get soft and thin from wear. If it does, much of the correction may be lost.
When the wedging method of shaping the cast is being used, check circulatory status frequently; it may be impaired by increased pressure on tissues and blood vessels. The equinus (posterior release) correction especially places considerable strain on ligaments, blood vessels, and tendons.
After surgery, elevate the child’s feet with pillows to decrease swelling and pain. Report signs of discomfort or pain right away. Try to locate the source of pain; it may result from cast pressure rather than from the incision. If bleeding occurs under the cast, circle the loca tion and mark the time on the cast. If bleeding spreads, report it.
Explain to the older child and his parents that surgery can improve clubfoot with good function but can’t totally correct it; the affected calf muscle will remain slightly underdeveloped.
Emphasize the need for long-term orthopedic care to maintain correction. Teach parents the prescribed exercises that the child can do at home. Urge them to make the child wear the corrective shoes ordered and the splints during naps and at night. Make sure they understand that treatment for clubfoot continues during the entire growth period. Correcting this defect permanently takes time and patience.
Developmental dysplasia of the hip
Developmental dysplasia of the hip (DDH), an abnormality of the hip joint present from birth, is the most common disorder affecting hip joints of children younger than age 3. DDH can be unilateral or bilateral. (See Characteristics of developmental hip dysplasia.) This abnormality occurs
in three forms of varying severity: unstable hip dysplasia, in which the hip is positioned normally but can be dislocated by manipulation; subluxati on or incomplete dislocation, in which the femoral head rides on the edge of the acetabulum; and complete or true congenital dislocation, in which the femoral head is totally outside the acetabulum.
in three forms of varying severity: unstable hip dysplasia, in which the hip is positioned normally but can be dislocated by manipulation; subluxati on or incomplete dislocation, in which the femoral head rides on the edge of the acetabulum; and complete or true congenital dislocation, in which the femoral head is totally outside the acetabulum.
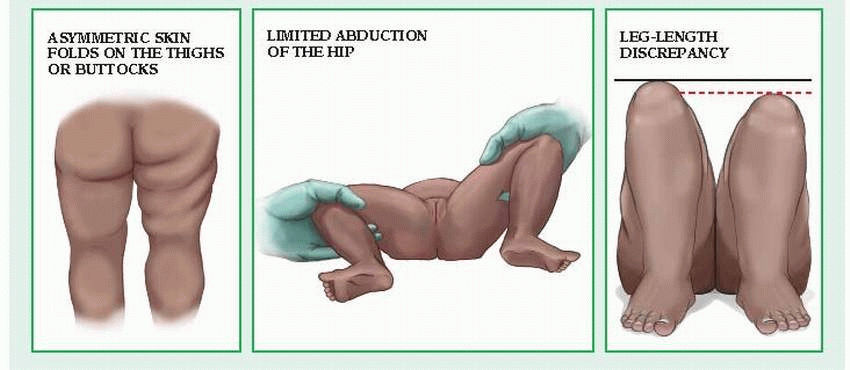
Developmental hip subluxation or dislocation can cause abnormal acetabular development and permanent disability.
CAUSES AND INCIDENCE
Experts are uncertain about the causes of DDH. Dislocation is 10 times more common after breech delivery (malpositioning in utero) than after cephalic delivery, and it’s also more common among large neonates and twins. It’s a lot more common in firstborn children. Girls are affected more often than boys and white children more than black children. Genetic factors may also play a role.
Although DDH is found throughout the world, incidence is particularly high among Native Americans.
COMPLICATIONS
Degenerative hip changes (if treatment is delayed)
Lordosis
Joint malformation
Crippling osteoarthritis
SIGNS AND SYMPTOMS
Clinical effects of hip dysplasia vary with age. In neonates, dysplasia doesn’t cause gross deformity or pain. However, in complete dysplasia, the hip rides above the acetabulum, causing the level of the knees to be uneven. As the child grows older and begins to walk, the abduction on the dislocated side is limited. Uncorrected bilateral dysplasia may cause him to sway from side to side, a condition known as “duck waddle”; unilateral dysplasia may produce a limp. If corrective treatment isn’t begun until after age 2, DDH may cause degenerative hip changes, lordosis, joint malformation, and softtissue damage.
DIAGNOSIS
Several observations during physical examination of the relaxed child strongly suggest DDH. First, place the child on his back, and inspect the folds of skin over his thighs. Usually, a child in this position has an equal number of thigh folds on each side, but a child with subluxation or dislocation may have an extra fold on the affected side (this extra fold is also apparent when the child lies prone). Next, with the child lying prone, check for alignment of the buttock fold. In a child with dysplasia, the buttock fold on the affected side is higher. In addition, abduction of the affected hip is restricted.
A positive Ortolani’s or Trendelenburg’s sign confirms DDH. To elicit Ortolani’s sign, place the infant on his back, with his hip flexed and abducted. Adducting the hip while pressing the femur downward will dislocate the hip. Then, abducting the hip while moving the femur upward will move the femoral head over the acetabular rim. If you hear a click or feel a jerk as the femoral head moves, the test is positive. This sign indicates subluxation in a neonate younger than 1 month and subluxation or complete dislocation in an older infant.
To elicit Trendelenburg’s sign, have the child rest his weight on the side of the dislocation and lift his other knee. His pelvis drops on the normal side because of weak abductor muscles in the affected hip. However, when the child stands with his weight on the normal side and lifts the other knee, the pelvis remains horizontal.
Ultrasound of the hip reveals hip deformity. X-rays show the location of the femur head and a shallow acetabulum. X-rays may also show acetabular dysplasia or a teratologica dislocation. Magnetic resonance imaging may also be used to assess reduction.
TREATMENT
The earlier the infant receives treatment, the better his chances are for normal development. Treatment varies with the patient’s age and is tailored to the specific pathological condition. In infants younger than 6 months, treatment includes gentle manipulation to reduce the dislocation, followed by holding the hips in a flexed and abducted position with a splint-brace or harness to maintain the reduction. The infant must wear this apparatus continuously for 2 to 3 months and then use a night splint for another month so the joint capsule can tighten and stabilize in correct alignment.
If treatment doesn’t begin until after age 3 months, it may include bilateral skin traction (in infants) or skeletal traction (in children who have started walking) in an attempt to reduce the dislocation by gradually abducting the hips. In Bryant’s traction, or divarication traction, both legs are placed in traction, even if only one is affected, to help maintain immobilization. This type of traction is used in children who are younger than 3 years and weigh less than 35 lb (16 kg). The length of treatment is 2 to 3 weeks.
If traction fails, gentle closed reduction under general anesthetic can further abduct the hips; the child is then placed in a spica cast for 4 to 6 months. If closed treatment fails, open reduction, followed by immobilization in a spica cast for an average of 6 months, or osteotomy may be considered.
In the child age 2 to 5 years, treatment is difficult and includes skeletal traction and subcutaneous adductor tenotomy. Treatment begun after age 5 rarely restores satisfactory hip function.
SPECIAL CONSIDERATIONS
The child who must wear a splint, brace, or body cast needs special personal care that requires parent education.
Teach parents how to correctly splint or brace the hips, as ordered. Stress the need for frequent checkups.
Listen sympathetically to the parents’ expressions of anxiety and fear. Explain possible causes of DDH, and give reassurance that early, prompt treatment will probably result in complete correction.
During the child’s first few days in a cast or splint-brace, he may be prone to irritability due to the unaccustomed restricted movement. Encourage his parents to stay with him as much as possible and to calm and reassure him.
Assure parents that the child will adjust to this restriction and return to normal sleeping, eating, and playing behavior in a few days.
Instruct parents to remove braces and splints while bathing the infant but to replace them immediately afterward. Stress good hygiene; parents should bathe and change the child frequently and wash his perineum with warm water and soap at each diaper change.
If treatment requires a spica cast:
When transferring the child immediately after casting, use your palms to avoid making dents in the cast. Such dents predispose the patient to pressure sores. Remember that the plaster cast needs 24 to 48 hours to dry naturally. Don’t use heat to make it dry faster because heat also makes it more fragile.
Immediately after the cast is applied, use a plastic sheet to protect it from moisture around the perineum and buttocks. Cut the sheet into strips long enough to cover the outside of the cast, and tuck them about a finger length beneath the cast edges. Using overlapping strips of tape, tack the corner of each petal to the outside of the cast. Remove the plastic under the cast every 4 hours; then wash, dry, and retuck it. Disposable diapers folded lengthwise over the perineum may also be used.
Position the child either on a Bradford frame elevated on blocks, with a bedpan under the frame, or on pillows to support the child’s legs. Be sure to keep the cast dry, and change the child’s diapers often.
Turn the child every 2 hours during the day and every 4 hours at night. Check color, sensation, and motion of the infant’s legs and feet. Be sure to examine all his toes. Notify the physician of dusky, cool, or numb toes.
Check the cast daily for odors, which may herald infection.
If the child complains of itching, he may benefit from diphenhydramine, or you may aim a hair dryer set on cool at the cast edges to relieve itching. Don’t scratch or probe under the cast. Investigate any persistent itching.
Provide adequate nutrition, and maintain adequate fluid intake to avoid renal calculi and constipation, both complications of inactivity.
Provide adequate stimuli to promote growth and development.
Tell parents to watch for signs that the child is outgrowing the cast (cyanosis, cool limbs, or pain).
Tell parents that treatment may be prolonged and requires patience.
The patient in Bryant’s traction may be cared for at home if the parents are taught traction application and maintenance.
Encourage the parents to cuddle and hold the child and encourage him to interact with sib lings and friends.
Maintain skin integrity and check circulation at least every 2 hours.
Feed the child carefully to avoid aspiration and choking.
Refer the child and his parents to a child life specialist to ensure continued developmental progress.
Muscular dystrophy
Muscular dystrophy is actually a group of congenital disorders characterized by progressive symmetrical wasting of skeletal muscles without neural or sensory defects. Paradoxically, these wasted muscles tend to enlarge because of connective tissue and fat deposits, giving an erroneous impression of muscle strength. The main types of muscular dystrophy are Duchenne’s (pseudohypertrophic), Becker’s (benign pseudohypertrophic), facioscapulohumeral (Landouzy-Dejerine), limb-girdle dystrophy,
Emery-Dreifuss muscular dystrophy, and myotonia congenita.
Emery-Dreifuss muscular dystrophy, and myotonia congenita.
The prognosis varies. Duchenne’s muscular dystrophy generally strikes during early childhood and usually results in death in the 20s or early 30s. Patients with Becker’s muscular dystrophy typically live into their 40s. Facioscapulohumeral and limb-girdle dystrophies usually don’t shorten life.
CAUSES AND INCIDENCE
Muscular dystrophy is caused by various genetic mechanisms. Duchenne’s and Becker’s muscular dystrophies are X-linked recessive disorders. Both result from defects in the gene coding for the muscle protein dystrophin; the gene has been mapped to the Xp21 locus.
The incidence of muscular dystrophy is about 1 in 651,450 persons in the United States. Duchenne’s and Becker’s muscular dystrophies affect males almost exclusively.
Facioscapulohumeral dystrophy is an autosomal dominant disorder. Limb-girdle dystrophy is usually autosomal recessive. These two types affect both sexes about equally.
COMPLICATIONS
Inhibited pulmonary function due to deformities
Greater risk for pneumonia
Respiratory problems lead to arrhythmias and hypertrophy
SIGNS AND SYMPTOMS
Although all four types of muscular dystrophy cause progressive muscular deterioration, the degree of severity and age of onset vary.
Duchenne’s muscular dystrophy begins insidiously, between ages 2 and 3. Initially, it affects leg and pelvic muscles but eventually spreads to the involuntary muscles. Muscle weakness produces a waddling gait, toe walking, and lordosis. Children with this disorder have difficulty climbing stairs, fall down often, can’t run properly, and their scapulae flare out (or “wing”) when they raise their arms. Calf muscles especially become enlarged and firm. Muscle deterioration progresses rapidly, and contractures develop. Some have abrupt intermittent oscillations of the irises in response to light (Gower’s sign). Usually, these children are confined to wheelchairs by ages 9 to 12. Late in the disease, progressive weakening of cardiac muscle causes tachycardia, electrocardiogram abnormalities, and pulmonary complications. Death commonly results from sudden heart failure, respiratory failure, or infection.
Signs and symptoms of Becker’s muscular dystrophy resemble those of Duchenne’s muscular dystrophy, but they progress more slowly. It generally affects older boys and young men. Children affected usually walk through their teens and into adulthood—sometimes into their 40s. Cardiac involvement is much less frequent.
Facioscapulohumeral dystrophy is a slowly progressive and relatively benign form of muscular dystrophy that commonly occurs before age 10 but may develop during early adolescence. The earlier the disease occurs, the more rapid and progressive it is. Initially, it weakens the muscles of the face, shoulders, and upper arms but eventually spreads to all voluntary muscles, producing a pendulous lower lip and absence of the nasolabial fold. Early symptoms include the inability to pucker the mouth or whistle, abnormal facial movements, and the absence of facial movements when laughing or crying. Other signs consist of diffuse facial flattening that leads to a masklike expression, winging of the scapulae, the inability to raise the arms above the head and, in infants, the inability to suckle.
Limb-girdle dystrophy follows a similarly slow course and commonly causes only slight disability. Usually, it begins between ages 6 and 10; less commonly, in early adulthood. The later the onset, the more rapid the progression. Muscle weakness first appears in the upper arm and pelvic muscles. Other symptoms include winging of the scapulae, lordosis with abdominal protrusion, waddling gait, poor balance, and the inability to raise the arms.
DIAGNOSIS
Diagnosis depends on typical clinical findings, family history, and diagnostic test findings. If another family member has muscular dystrophy, its clinical characteristics can indicate the type of dystrophy the patient has and how he may be affected.
Electromyography typically demonstrates short, weak bursts of electrical activity or high-frequency, repetitive waxing and waning discharges in affected muscles. Muscle biopsy shows variations in the size of muscle fibers and, in later stages, shows fat and connective tissue deposits; dystrophin is absent in Duchenne’s dystrophy and diminished in Becker’s dystrophy. Serum creatine kinase level is markedly elevated in Duchenne’s, but only moderately elevated in Becker’s and facioscapulohumeral dystrophies.
Immunologic and molecular biological assays available in specialized medical centers
facilitate accurate prenatal and postnatal diagnosis of Duchenne’s and Becker’s muscular dystrophies and are replacing muscle biopsy and elevated serum creatine kinase levels in diagnosing these dystrophies. These assays can also help to identify carriers.
facilitate accurate prenatal and postnatal diagnosis of Duchenne’s and Becker’s muscular dystrophies and are replacing muscle biopsy and elevated serum creatine kinase levels in diagnosing these dystrophies. These assays can also help to identify carriers.
TREATMENT
No treatment stops the progressive muscle impairment of muscular dystrophy. However, orthopedic appliances, exercise, physical therapy, and surgery to correct contractures can help preserve the patient’s mobility and independence. Prednisone improves muscle strength in patients with Duchenne’s.
SPECIAL CONSIDERATIONS
Comprehensive long-term care and follow-up, patient and family teaching, and psychological support can help the patient and his family deal with this disorder.
When respiratory involvement occurs in Duchenne’s muscular dystrophy, encourage coughing, deep-breathing exercises, and diaphragmatic breathing. Teach parents how to recognize early signs of respiratory complications.
Encourage and assist with active and passive range-of-motion exercises to preserve joint mobility and prevent muscle atrophy.
Advise the patient to avoid long periods of bed rest and inactivity; if necessary, limit TV viewing and other sedentary activities.
Refer the patient for physical therapy. Splints, braces, trapeze bars, overhead slings, and a wheelchair can help preserve mobility. A footboard or high-topped sneakers and a foot cradle increase comfort and prevent footdrop.
Refer the patient to surgery to correct contractures.
Because inactivity may cause constipation, encourage adequate fluid intake, increase dietary bulk, and obtain an order for a stool softener. The patient is prone to obesity due to reduced physical activity; help him and his family plan a low-calorie, high-protein, highfiber diet.
Always allow the patient plenty of time to perform even simple physical tasks because he’s likely to be slow and awkward.
Encourage communication between the patient’s family members to help them deal with the emotional strain this disorder produces. Provide emotional support to help the patient cope with continual changes in body image.
Help the child with Duch enne’s muscular dystrophy to maintain peer relationships and to realize his intellectual potential by encouraging his parents to keep him in a regular school as long as possible.
If necessary, refer adult patients for counseling. Refer those who must acquire new job skills for vocational rehabilitation. (Contact the Department of Labor and Industry in your state for more information.) For information on social services and financial assistance, refer these patients and their families to the Muscular Dystrophy Association.
Refer the patient’s family members for genetic counseling.
JOINTS
Septic arthritis
Septic, or infectious, arthritis is a medical emergency that occurs when bacterial invasion of a joint causes inflammation of the synovial lining, effusion and pyogenesis, and destruction of bone and cartilage. Septic arthritis can lead to ankylosis and even fatal septicemia. However, prompt antibiotic therapy and joint aspiration or drainage cures most patients.
CAUSES AND INCIDENCE
In most cases of septic arthritis, bacteria spread from a primary site of infection—usually in adjacent bone or soft tissue—through the bloodstream to the joint. Common infecting organisms in children are group B Streptococcus and Haemophilus influenzae. Adults are usually infected by Staphylococcus, Streptococcus, Neisseria gonorrhoeae (pneumonia), and group B Streptococcus, whereas chronic septic arthritis is caused by Mycobacterium tuberculosis and Candida albicans.
Various factors can predispose a person to septic arthritis. Any concurrent bacterial infection (of the genitourinary or the upper respiratory tract, for example) or serious chronic illness (such as malignancy, renal failure, rheumatoid arthritis, systemic lupus erythematosus, diabetes, or cirrhosis) heightens susceptibility. Consequently, elderly people and those who abuse I.V. drugs run a higher risk of developing septic arthritis. Of course, diseases that depress the immune system and immunosuppressant therapy increase susceptibility. Other predisposing factors include recent articular trauma, joint arthroscopy or other surgery, intra-articular injections, local joint abnormalities, animal or human bites, and nail puncture wounds.
Other types of arthritis
Hemophilic arthrosis
Hemophilic arthrosis produces transient or permanent joint changes. Often precipitated by trauma, hemophilic arthrosis usually arises between ages 1 and 5 and tends to recur until about age 10. It usually affects only one joint at a time—most commonly the knee, elbow, or ankle—and tends to recur in the same joint. Initially, the patient may feel only mild discomfort; later, he may experience warmth, swelling, tenderness, and severe pain with adjacent muscle spasm that leads to flexion of the extremity.
Mild hemophilic arthrosis may cause only limited stiffness that subsides within a few days. In prolonged bleeding, however, symptoms may subside after weeks or months or not at all. Severe hemophilic arthrosis may be accompanied by fever and leukocytosis; severe, prolonged, or repeated bleeding may lead to chronic hemophilic joint disease.
Effective treatment includes I.V. infusion of the deficient clotting factor, bed rest with the affected extremity elevated, application of ice packs, analgesics, and joint aspiration. Physical therapy includes progressive range-of-motion and muscle-strengthening exercises to restore motion and to prevent contractures and muscle atrophy.
Intermittent hydrarthrosis
Intermittent hydrarthrosis is a rare, benign condition characterized by regular, recurrent joint effusions. It most commonly affects the knee. The patient may have difficulty moving the affected joint but have no other arthritic symptoms. The cause of intermittent hydrarthrosis is unknown; onset is usually at or soon after puberty and may be linked to familial tendencies, allergies, or menstruation. No effective treatment exists.
Henoch-Schönlein purpura
Henoch-Schönlein purpura—a vasculitic syndrome—is marked by palpable purpura, abdominal pain, and arthralgia that most commonly affects the knees and ankles, producing swollen, warm, and tender joints without joint erosion or deformity. Renal involvement is also common. Most patients have microscopic hematuria and proteinuria 4 to 8 weeks after onset. Incidence is highest in children and young adults, occurring most often in the spring after a respiratory infection. Treatment may include corticosteroids.
Traumatic arthritis
Traumatic arthritis results from blunt, penetrating, or repeated trauma or from forced inappropriate motion of a joint or ligament. Clinical effects may include swelling, pain, tenderness, joint instability, and internal bleeding. Treatment includes analgesics, nonsteroidal anti-inflammatory drugs, application of cold followed by heat and, if needed, compression dressings, splinting, joint aspiration, casting, or possibly surgery.
Septic arthritis may be seen at any age in children, but it occurs most often in children younger than age 3. It’s uncommon from age 3 until adolescence, at which time the incidence increases again.
COMPLICATIONS
Joint degeneration
Osteomyelitis
SIGNS AND SYMPTOMS
Acute septic arthritis begins abruptly, causing intense pain, inflammation, and swelling of the affected joint and low-grade fever. It usually affects a single joint. It most commonly develops in the large joints but can strike any joint, including the spine and small peripheral joints. The hip is a frequent site in infants. Systemic signs of inflammation may not appear in some patients. Migratory polyarthritis sometimes precedes localization of the infection. If the bacteria invade the hip, pain may occur in the groin, upper thigh, or buttock or may be referred to the knee.
DIAGNOSIS
Identifying the causative organism in a Gram stain or culture of synovial fluid or a biopsy of synovial membrane confirms septic arthritis. When synovial fluid culture is negative, positive blood culture may confirm the diagnosis. Ultrasound of the hip is the modality of choice to detect fluid collections in the hip joint and can serve as a guide during aspiration procedures.
Joint fluid analysis shows gross pus or watery, cloudy fluid of decreased viscosity, usually with 50,000/µl or more white cells, primarily neutrophils. Synovial fluid glucose concentration is usually greater than 40 mg/dl. (See Other types of arthritis.)
Other diagnostic measures include the following:
X-rays can show typical changes as early as 1 week after initial infection—distention of joint capsules, for example, followed by narrowing of joint space (indicating cartilage damage) and erosions of bone (joint destruction).
White blood cell count may be elevated, with many polymorphonuclear cells; erythrocyte sedimentation rate is increased.
Triple-phase bone scan is often used in children. A whole body scan is preferred in very young children.
CT and MRI can provide useful images to delineate the extent of the infection.
TREATMENT
Antibiotic therapy should begin as soon as a Gram stain has been done; it may be modified when drug sensitivity of the infecting organism is known. Bioassays or bactericidal assays of synovial fluid and bioassays of blood may confirm clearing of the infection.
Rest, immobilization, elevation, and warm compresses help with pain relief. Analgesics are given for pain, if needed. The affected joint can be immobilized with a splint or put into traction until the patient can tolerate movement.
In severe cases, needle aspiration (arthrocentesis) or surgery may be done under sterile conditions to remove grossly purulent or infected joint fluid. Late reconstructive surgery is warranted only for severe joint damage and only after all signs of active infection have disappeared, which usually takes several months. Recommended procedures include arthroplasty and joint fusion. Prosthetic replacement remains controversial because it may exacerbate the infection, but it has helped patients with damaged femoral heads or acetabula.
SPECIAL CONSIDERATIONS
Management of septic arthritis demands meticulous supportive care, close observation, and control of infection.
Practice strict sterile technique for all procedures. Wash hands carefully before and after giving care. Dispose of soiled linens and dressings properly. Prevent contact between immunosuppressed patients and infected patients.
Watch for signs of joint inflammation: heat, redness, swelling, pain, or drainage. Monitor vital signs and fever pattern. Remember that corticosteroids mask signs of infection.
Check splints or traction regularly. Keep the joint in proper alignment, but avoid prolonged immobilization. Start passive range-of-motion exercises immediately, and progress to active exercises as soon as the patient can move the affected joint and put weight on it.
Monitor pain levels and medicate accordingly, especially before exercise, remembering that the pain of septic arthritis is easy to underestimate. Administer analgesics and opioids for acute pain and heat or ice packs for moderate pain.
Monitor older adults who are on long-term opioid therapy because these drugs can impair mental status and may contribute to falls and other accidents.
Warn the patient before the first aspira tion that it will be extremely painful. Carefully evaluate the patient’s condition after joint aspiration.
Gout
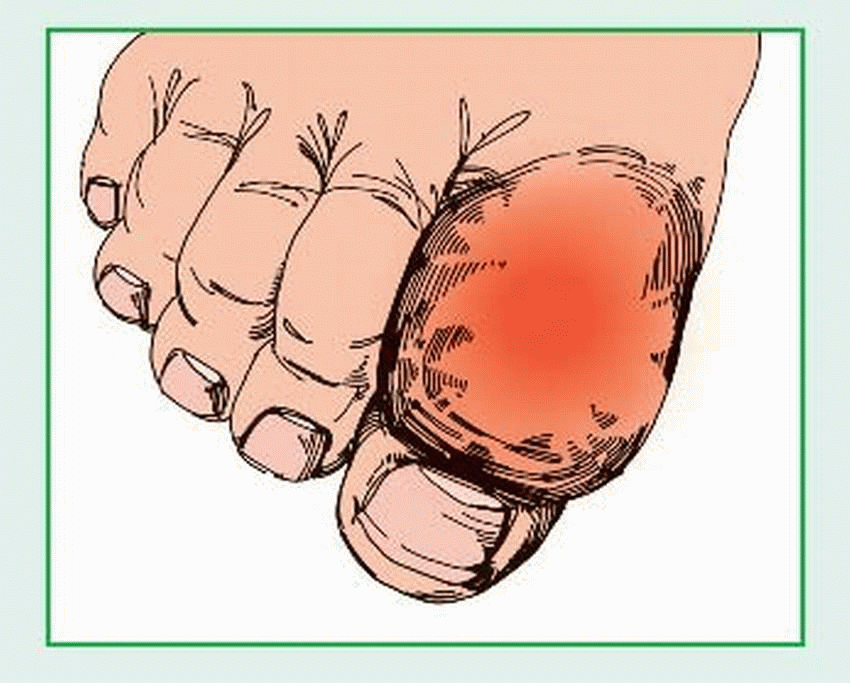
Gout, also called gouty arthritis, is a metabolic disease marked by urate deposits, which cause painfully arthritic joints. (See Gouty deposits, page 308.) It can strike any joint but favors those in the feet and legs. Gout follows an intermittent course and typically leaves patients totally free from symptoms for years between attacks. It can cause chronic disability or incapacitation and, rarely, severe hypertension and progressive renal disease. The prognosis is good with treatment.
CAUSES AND INCIDENCE
Although the exact cause of primary gout remains unknown, it appears to be linked to a genetic defect in purine metabolism, which causes elevated blood levels of uric acid (hyperuricemia) due to overproduction of uric acid, retention of uric acid, or both. In secondary gout, which develops during the course of another disease (such as obesity, diabetes mellitus, hypertension, sickle cell anemia, and renal disease), hyperuricemia results from the breakdown of nucleic acids. Myeloproliferative and lymphoproliferative diseases, psoriasis, and hemolytic anemia are the most common causes. Primary gout usually occurs in men and in postmenopausal women; secondary gout occurs in elderly people.
Secondary gout can also follow drug therapy that interferes with uric acid excretion. Increased concentration of uric acid leads to urate deposits (tophi) in joints or tissues and
consequent local necrosis or fibrosis. The risk is greater in men, postmenopausal women, and those who use alcohol.
consequent local necrosis or fibrosis. The risk is greater in men, postmenopausal women, and those who use alcohol.
COMPLICATIONS
Renal calculi
Atherosclerotic disease
Cardiovascular disease
Stroke
Coronary thrombosis
Hypertension
Infection when tophi rupture
SIGNS AND SYMPTOMS
Gout develops in four stages: asymptomatic, acute, intercritical, and chronic. In asymptomatic gout, serum urate levels rise but produce no symptoms. As the disease progresses, it may cause hypertension or nephrolithiasis, with severe back pain. The first acute attack strikes suddenly and peaks quickly. Although it generally involves only one or a few joints, this initial attack is extremely painful. Affected joints are hot, tender, inflamed, and appear dusky-red or cyanotic. The metatarsophalangeal joint of the great toe usually becomes inflamed first (podagra), followed by the instep, ankle, heel, knee, or wrist joints. Sometimes a low-grade fever is present. Mild acute attacks usually subside quickly but tend to recur at irregular intervals. Severe attacks may persist for days or weeks.
Intercritical periods are the symptom-free intervals between gout attacks. Most patients have a second attack within 6 months to 2 years, but in some the second attack doesn’t occur for 5 to 10 years. Delayed attacks are more common in untreated patients and tend to be longer and more severe than initial attacks. Such attacks are also polyarticular, invariably affecting joints in the feet and legs, and are sometimes accompanied by fever. A migratory attack sequentially strikes various joints and the Achilles tendon and is associated with either subdeltoid or olecranon bursitis.
Eventually, chronic polyarticular gout sets in. This final, unremitting stage of the disease is marked by persistent painful polyarthritis, with large, subcutaneous tophi in cartilage, synovial membranes, tendons, and soft tissue. Tophi form in fingers, hands, knees, feet, ulnar sides of the forearms, helix of the ear, Achilles tendons and, rarely, internal organs, such as the kidneys and myocardium. The skin over the tophus may ulcerate and release a chalky, white exudate or pus. Chronic inflammation and tophaceous deposits precipitate secondary joint degeneration, with eventual erosions, deformity, and disability. Kidney involvement, with associated tubular damage, leads to chronic renal dysfunction. Hypertension and albuminuria occur in some patients; urolithiasis is common.
DIAGNOSIS
The presence of monosodium urate monohydrate crystals in synovial fluid taken from an inflamed joint or tophus establishes the diagnosis.
Aspiration of synovial fluid (arthrocentesis) or of tophaceous material reveals needlelike intracellular crystals of sodium urate. Although hyperuricemia isn’t specifically diagnostic of gout, serum uric acid is above normal. Urinary uric acid is usually higher in secondary gout than in primary gout. In acute attacks, erythrocyte sedimentation rate and white blood cell (WBC) count may be elevated, and WBC count shifts to the left.
Initially, X-rays are normal. However, in chronic gout, X-rays show “punched out” erosions, sometimes with periosteal overgrowth. Outward displacement of the overhanging margin from the bone contour characterizes gout. X-rays rarely show tophi. (See Understanding pseudogout.)
TREATMENT
Correct management seeks to terminate an acute attack, reduce hyperuricemia, and prevent
recurrence, complications, and the formation of renal calculi. (See Preventing gout, page 310.) Colchicine is effective in reducing pain, swelling, and inflammation; pain often subsides within 12 hours of treatment and is completely relieved in 48 hours. Treatment for the patient with acute gout consists of bed rest; immobilization and protection of the inflamed, painful joints; and local application of heat or cold, whichever works for the patient. Maximal doses of nonsteroidal anti-inflammatory drugs (NSAIDs) usually provide excellent relief for patients who can tolerate them; doses should be gradually reduced after several days.
recurrence, complications, and the formation of renal calculi. (See Preventing gout, page 310.) Colchicine is effective in reducing pain, swelling, and inflammation; pain often subsides within 12 hours of treatment and is completely relieved in 48 hours. Treatment for the patient with acute gout consists of bed rest; immobilization and protection of the inflamed, painful joints; and local application of heat or cold, whichever works for the patient. Maximal doses of nonsteroidal anti-inflammatory drugs (NSAIDs) usually provide excellent relief for patients who can tolerate them; doses should be gradually reduced after several days.
Older patients are at risk for GI bleeding associated with NSAID use. Encourage the elderly patient to take these drugs with meals, and monitor the patient’s stools for occult blood.
Resistant inflammation may require oral corticosteroids or intra-articular corticosteroid injection to relieve pain. Treatment for chronic gout aims to decrease serum uric acid level. Continuing maintenance dosage of allopurinol may be given to suppress uric acid formation or control uric acid levels, preventing further attacks. However, this powerful drug should be used cautiously in patients with renal failure. Uricosuric agents promote uric acid excretion and inhibit accumulation of uric acid, but their value is limited in patients with renal impairment. These medications shouldn’t be given to patients with renal calculi.
Adjunctive therapy emphasizes a few dietary restrictions, primarily the avoidance of alcohol and purine-rich foods (organ meats, beer, wine, and certain types of fish are high in purines). Obese patients should try to lose weight because obesity puts additional stress on painful joints.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


