Endocrine Disorders
INTRODUCTION
Together with the nervous system, the endocrine system regulates and integrates the body’s metabolic activities. The endocrine system meets the nervous system at the hypothalamus. The hypothalamus, the main integrative center for the endocrine and autonomic nervous systems, controls the function of endocrine organs by neural and hormonal pathways. A hormone is a chemical transmitter released from specialized cells into the bloodstream, which carries it to specialized organ-receptor cells that respond to it.
Neural pathways connect the hypothalamus to the posterior pituitary, or neurohypophysis. Neural stimulation to the posterior pituitary provokes the secretion of two effector hormones: antidiuretic hormone (ADH) and oxytocin, and influences thyroid-stimulating hormone (TSH), corticotropin, prolactin, and gonadotropinreleasing hormone.
HYPOTHALAMIC CONTROL
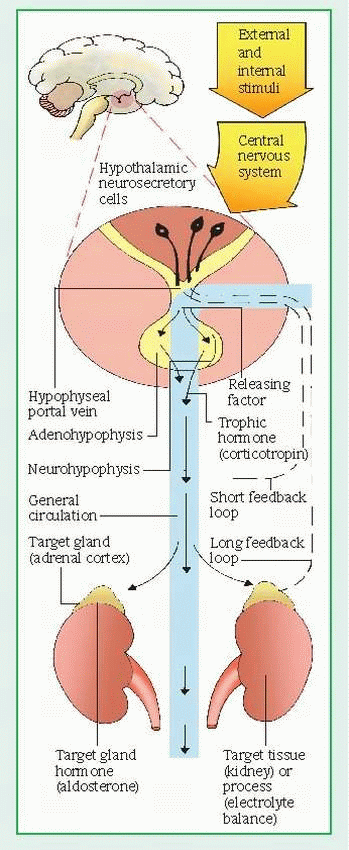
The hypothalamus also exerts hormonal control at the anterior pituitary through releasing and inhibiting factors, which arrive by a portal system. Hypothalamic hormones stimulate the pituitary to release trophic hormones, such as corticotropin, TSH, luteinizing hormone (LH), and follicle-stimulating hormone (FSH), and to release or inhibit effector hormones, such as the growth hormone and prolactin. In turn, secretion of trophic hormones stimulates the adrenal cortex, thyroid, and gonads. In a patient whose clinical condition suggests endocrine pathology, this complex hormonal sequence requires careful evaluation at each level to identify the dysfunction; dysfunction may result from defects of releasing, trophic, or effector hormones or of the target tissue. Hyperthyroidism, for example, may result from an excess of thyrotropinreleasing hormone, TSH, or thyroid hormone.
In addition to hormonal and neural controls, a negative feedback system regulates the endocrine system. (See Feedback mechanism of the endocrine system, page 552.) The mechanism of feedback may be simple or complex. Simple feedback occurs when the level of one substance regulates secretion of a hormone. For example, low serum calcium levels stimulate parathyroid hormone (PTH) secretion; high serum calcium levels inhibit it. Complex feedback occurs through the hypothalamic-pituitarytarget organ axis; for example, secretion of the hypothalamic corticotropin-releasing hormone (CRH) releases pituitary corticotropin, which, in turn, stimulates adrenal cortisol secretion. Subsequently, a rise in serum cortisol level inhibits corticotropin by decreasing CRH secretion. Steroid therapy disrupts the hypothalamic-pituitary-adrenal (HPA) axis by suppressing hypothalamic-pituitary secretion. Because abrupt withdrawal of steroids doesn’t allow time for recovery of the HPA axis to stimulate cortisol secretion, it can induce a life-threatening adrenal crisis.
HORMONAL EFFECTS
In response to the hypothalamus, the posterior pituitary secretes oxytocin and ADH. Oxytocin stimulates contraction of the uterus and is responsible for the milk let-down reflex in lactating females. ADH controls the concentration of body fluids by altering the permeability of the distal convoluted tubules and collecting ducts of the kidneys to conserve water. The secretion of ADH depends on plasma volume and osmolality as monitored by hypothalamic neurons. Circulatory shock and severe hemorrhage are the most powerful stimulators of ADH; other stimulators include pain, emotional stress, trauma, morphine, tranquilizers, certain anesthetics, and positive-pressure breathing.
The syndrome of inappropriate ADH secretion is a disorder that produces hyponatremia with water overload. Generally, however, overhydration suppresses ADH secretion (as does alcohol). ADH deficiency causes diabetes insipidus, a condition of high urine output.
The anterior pituitary secretes prolactin, which stimulates milk production, and human growth hormone (hGH), which stimulates growth by increasing protein synthesis and fat mobilization and by decreasing carbohydrate utilization. Hyposecretion of hGH results in dwarfism; hypersecretion causes gigantism in children and acromegaly in adults.
The thyroid gland secretes the iodinated hormones thyroxine and triiodothyronine. Thyroid hormones, necessary for normal growth and development, act on many tissues to increase metabolic activity and protein synthesis. Deficiency of thyroid hormone causes varying degrees of hypothyroidism, from a mild, clinically insignificant form to life-threatening myxedema coma. Congenital hypothyroidism causes cretinism. Hypersecretion causes hyperthyroidism and, in extreme cases, thyrotoxic crisis. Excessive secretion of TSH causes thyroid gland hyperplasia, resulting in goiter.
The parathyroid glands secrete PTH, which regulates calcium and phosphate metabolism. PTH elevates serum calcium levels by
stimulating resorption of calcium and phosphate from bone, reabsorption of calcium and excretion of phosphate by the kidneys and, by combined action with vitamin D, absorption of calcium and phosphate from the GI tract. PTH also stimulates conversion of vitamin D to its metabolically active form. Thyrocalcitonin, a secretion from the thyroid, opposes the effect of PTH and therefore decreases serum calcium levels. Hyperparathyroidism results in hypercalcemia, and hypoparathyroidism causes hypocalcemia. Altered calcium levels may also result from nonendocrine causes such as metastatic bone disease or nutritional vitamin D deficiency.
stimulating resorption of calcium and phosphate from bone, reabsorption of calcium and excretion of phosphate by the kidneys and, by combined action with vitamin D, absorption of calcium and phosphate from the GI tract. PTH also stimulates conversion of vitamin D to its metabolically active form. Thyrocalcitonin, a secretion from the thyroid, opposes the effect of PTH and therefore decreases serum calcium levels. Hyperparathyroidism results in hypercalcemia, and hypoparathyroidism causes hypocalcemia. Altered calcium levels may also result from nonendocrine causes such as metastatic bone disease or nutritional vitamin D deficiency.
The endocrine part of the pancreas produces glucagon from the alpha cells and insulin from the beta cells. Glucagon, the hormone of the fasting state, releases stored glucose to raise the blood glucose level. Insulin, the hormone of the nourished state, facilitates glucose transport, promotes glucose storage, stimulates protein synthesis, and enhances free fatty acid uptake and storage. Absolute or relative insulin deficiency causes diabetes mellitus. Insulin excess can result from an insulinoma (a tumor of the beta cells).
A common and important endocrine change in older people is a decreased ability to tolerate stress, as demonstrated by glucose metabolism. Normally, fasting blood glucose levels aren’t significantly different in young and old adults. However, when stress stimulates an older person’s pancreas, the blood glucose concentration increases and remains elevated longer than in a young adult.
The adrenal cortex secretes mineralocorticoids, glucocorticoids, and sex steroids. Aldosterone, a mineralocorticoid, regulates the reabsorption of sodium and the excretion of potassium by the kidneys. Although affected by corticotropin, aldosterone is regulated by angiotensin II, which in turn, is regulated by renin and plasma volume. Together, aldosterone, angiotensin, and renin may be implicated in the pathogenesis of hypertension. An excess of aldosterone (aldosteronism) can result primarily from hyperplasia or from adrenal adenoma or secondarily from many conditions, including heart failure and cirrhosis.
Cortisol, a glucocorticoid, stimulates gluconeogenesis, increases protein breakdown and free fatty acid mobilization, suppresses the immune response, and provides for an appropriate response to stress. Hyperactivity of the adrenal cortex results in Cushing’s syndrome; hypoactivity of the adrenal cortex causes Addison’s disease and, in extreme cases, adrenal crisis. Adrenogenital syndromes may result from overproduction of sex steroids.
The adrenal medulla is an aggregate of nervous tissue that produces the catecholamines epinephrine and norepinephrine, both of which cause vasoconstriction. Epinephrine also causes
the fight-or-flight response—dilation of bronchioles and increased blood pressure, blood glucose levels, and heart rate. Pheochromocytoma, a tumor of the adrenal medulla, causes hypersecretion of catecholamines and results in characteristic sustained or paroxysmal hypertension. The testes synthesize and secrete testosterone in response to gonadotropic hormones, especially LH, from the anterior pituitary gland; spermatogenesis occurs in response to FSH. The ovaries produce sex steroid hormones, primarily estrogen and progesterone, in response to LH and FSH.
the fight-or-flight response—dilation of bronchioles and increased blood pressure, blood glucose levels, and heart rate. Pheochromocytoma, a tumor of the adrenal medulla, causes hypersecretion of catecholamines and results in characteristic sustained or paroxysmal hypertension. The testes synthesize and secrete testosterone in response to gonadotropic hormones, especially LH, from the anterior pituitary gland; spermatogenesis occurs in response to FSH. The ovaries produce sex steroid hormones, primarily estrogen and progesterone, in response to LH and FSH.
ENDOCRINE DYSFUNCTION
Chronic endocrine abnormalities are common health problems. For example, deficiencies of cortisol, thyroid hormone, or insulin may require lifelong hormone replacement for survival. Consequently, these conditions make special demands on your skills during ongoing patient assessment, management of acute illness, and patient teaching.
Common dysfunctions of the endocrine system are classified as hypofunction and hyperfunction, inflammation, and tumor. The source of hypofunction and hyperfunction may originate in the hypothalamus or in the pituitary or effector glands. Inflammation may be acute or subacute, as in thyroiditis, but is usually chronic, commonly resulting in glandular hypofunction. Tumors can occur within a gland—as in thyroid cancer or adrenal pheochromocytoma—or in other areas, resulting in ectopic hormone production. Certain lung tumors, for example, secrete ADH, PTH, or structurally similar substances that have the same effects on target tissues.
The study of endocrine function focuses on measuring the level or effect of a hormone. Radioimmunoassay, for example, measures insulin levels; a fasting blood glucose test measures insulin’s effects. Sophisticated techniques of hormone measurement have improved diagnosis of endocrine disorders.
Diagnostic tests confirm endocrine disorders, but clinical data usually provide the first clues. Nursing assessment can reveal such signs and symptoms as excessive or delayed growth, wasting, weakness, polydipsia, polyuria, and mental changes. The quality and distribution of hair, skin pigmentation, and the distribution of body fat are also significant.
Nurses are also responsible for patient preparation, including instruction and support during testing, and for specimen collection, particularly of timed blood and urine specimens.
PITUITARY GLAND
Hypopituitarism
Hypopituitarism, which includes panhypopituitarism and dwarfism, is a complex syndrome marked by metabolic dysfunction, sexual immaturity, and growth retardation (when it occurs in childhood), resulting from a deficiency of the hormones secreted by the anterior pituitary gland. Panhypopituitarism refers to a generalized condition caused by partial or total failure of the anterior pituitary’s vital hormones—corticotropin, thyroid-stimulating hormone (TSH), luteinizing hormone (LH), follicle-stimulating hormone (FSH), human growth hormone (hGH), and prolactin—plus the posterior pituitary hormone, antidiuretic hormone. Partial hypopituitarism and complete hypopituitarism occur in adults and children; in children, these diseases may cause dwarfism and delayed puberty. The prognosis may be good with adequate replacement therapy and correction of the underlying causes.
CAUSES AND INCIDENCE
The most common cause of primary hypopituitarism in adults is a tumor. Other causes include congenital defects (hypoplasia or aplasia of the pituitary gland); pituitary infarction (most often from postpartum hemorrhage); or partial or total hypophysectomy by surgery, irradiation, or chemical agents; and, rarely, granulomatous disease (e.g., tuberculosis). Occasionally, hypopituitarism may have no identifiable cause, or it may be related to autoimmune destruction of the gland. Secondary hypopituitarism stems from a deficiency of releasing hormones produced by the hypothalamus—either idiopathic or possibly resulting from infection, trauma, or a tumor.
Primary hypopituitarism usually develops in a predictable pattern of hormonal failures. It generally starts with hypogonadism from gonadotropin failure (decreased FSH and LH levels). In adults, it causes cessation of menses in females and impotence in men. Growth hormone (GH) deficiency follows; in children, this causes short stature, delayed growth, and delayed puberty. In adults, it causes osteoporosis, decreased leanto-fat body mass index, adverse lipid changes, and subtle emotional dysphoria and lethargy. Subsequent failure of thyrotropin (decreased TSH levels) causes hypothyroidism; finally, adrenocorticotropic failure (decreased corticotropin levels) results in adrenal insufficiency. However,
when hypopituitarism follows surgical ablation or trauma, the pattern of hormonal events may not necessarily follow this sequence. Sometimes, damage to the hypothalamus or neurohypophysis from one of the above leads to diabetes insipidus. Hypopituitarism may develop years after pituitary radiation treatment.
when hypopituitarism follows surgical ablation or trauma, the pattern of hormonal events may not necessarily follow this sequence. Sometimes, damage to the hypothalamus or neurohypophysis from one of the above leads to diabetes insipidus. Hypopituitarism may develop years after pituitary radiation treatment.
SIGNS AND SYMPTOMS
Clinical features of hypopituitarism develop slowly and vary with the severity of the disorder and the number of deficient hormones. Signs and symptoms of hypopituitarism in adults may include gonadal failure (secondary amenorrhea, impotence, infertility, decreased libido), diabetes insipidus, hypothyroidism (fatigue, lethargy, sensitivity to cold, menstrual disturbances), and adrenocortical insufficiency (hypoglycemia, anorexia, nausea, abdominal pain, orthostatic hypotension).
Postpartum necrosis of the pituitary (Sheehan’s syndrome) characteristically causes failure of lactation, menstruation, and growth of pubic and axillary hair; and symptoms of thyroid and adrenocortical failure.
In children, hypopituitarism causes retarded growth or delayed puberty. Dwarfism usually isn’t apparent at birth but early signs begin to appear during the first few months of life; by age 6 months, growth retardation is obvious. Although these children generally enjoy good health, pituitary dwarfism may cause chubbiness due to fat deposits in the lower trunk, delayed secondary tooth eruption and, possibly, hypoglycemia. Growth continues at less than half the normal rate—sometimes extending into the patient’s 20s or 30s—to an average height of 4′ (122 cm), with normal proportions.
When hypopituitarism strikes before puberty, it prevents development of secondary sex characteristics (including facial and body hair). In males, it produces undersized testes, penis, and prostate gland; absent or minimal libido; and the inability to initiate and maintain an erection. In females, it usually causes immature development of the breasts, sparse or absent pubic and axillary hair, and primary amenorrhea.
Panhypopituitarism may induce a host of mental and physiologic abnormalities, including lethargy, psychosis, orthostatic hypotension, bradycardia, anemia, and anorexia. However, clinical manifestations of hormonal deficiencies resulting from pituitary destruction don’t become apparent until 75% of the gland is destroyed. Total loss of all hormones released by the anterior pituitary is fatal unless treated.
Neurologic signs associated with hypopituitarism and produced by pituitary tumors include headache, bilateral temporal hemianopia, loss of visual acuity and, possibly, blindness. Acute hypopituitarism resulting from surgery or infection is often associated with fever, hypotension, vomiting, and hypoglycemia—all characteristic of adrenal insufficiency.
DIAGNOSIS
In suspected hypopituitarism, evaluation must confirm hormonal deficiency due to impairment or destruction of the anterior pituitary gland and rule out disease of the target organs (adrenals, gonads, and thyroid) or the hypothalamus. Low serum levels of thyroxine (T4), for example, indicate diminished thyroid gland function, but further tests are necessary to identify the source of this dysfunction as the thyroid, pituitary, or hypothalamus.
Serum insulin-like growth factor 1 (IGF-1) is decreased. Cranial computed tomography (CT) scan or magnetic resonance imaging (MRI) may reveal a tumor or abnormal mass in the pituitary gland or the hypothalamus. Radioimmunoassay showing decreased plasma levels of some or all pituitary hormones, accompanied by end-organ hypofunction, suggests pituitary failure, and eliminates target gland disease. Failure of thyrotropin-releasing hormone administration to increase TSH or prolactin concentrations rules out hypothalamic dysfunction as the cause of hormonal deficiency.
Provocative tests are helpful in pinpointing the source of low cortisol levels. Oral metyrapone blocks cortisol synthesis, which should stimulate pituitary secretion of corticotropin and the adrenal precursors of cortisol, measured in urine as hydroxycorticosteroids. Insulin-induced hypoglycemia also stimulates corticotropin secretion. Persistently low levels of corticotropin indicate pituitary or hypothalamic failure. These tests require careful medical supervision because they may precipitate an adrenal crisis.
Diagnosis of hypopituitarism requires measurement of GH levels in the blood after administration of regular insulin (inducing hypoglycemia) or levodopa (causing hypotension). These drugs should provoke increased secretion of GH. Persistently low GH levels, despite provocative testing, confirm GH deficiency. CT scan, MRI, or cerebral angiography confirms the presence of intrasellar or extrasellar tumors.
TREATMENT
Replacement of hormones secreted by the target glands is the most effective treatment for hypopituitarism. Hormone replacement therapy includes cortisol, T4, and androgen or cyclic estrogen. Prolactin need not be replaced. The patient of reproductive age may benefit from administration of FSH and human chorionic gonadotropin to boost fertility. GH replacement is recommended for adults as well as children. Replacement is done by administering daily subcutaneous injections of one of several recombinant deoxyribonucleic acid (DNA) GHs, accompanied by follow-up of serum IGF-1 levels. Lean body mass increases, whereas adipose tissue—particularly in the abdomen—decreases. Risk of cardiovascular disease and osteoporosis also decrease with treatment. Many patients also notice an improved sense of well-being.
Somatrem, which is identical to hGH but is the product of recombinant DNA technology, has replaced GHs derived from human sources. It’s effective for treating dwarfism and stimulates growth increases as great as 4” to 6” (10 to 15 cm) in the first year of treatment. The growth rate tapers off in subsequent years. After pubertal changes have occurred, the effects of somatrem therapy are limited. Occasionally, a child becomes unresponsive to somatrem therapy, even with larger doses, perhaps because antibodies have formed against it. In such refractory patients, small doses of androgen may again stimulate growth, but extreme caution is necessary to prevent premature closure of the epiphyses. Children with hypopituitarism may also need replacement of adrenal and thyroid hormones and, as they approach puberty, sex hormones.
SPECIAL CONSIDERATIONS
Caring for patients with hypopituitarism requires an understanding of hormonal effects and skilled physical and psychological support.
Monitor the results of all laboratory tests for hormonal deficiencies, and know what they mean. Until hormone replacement therapy is complete, check for signs of thyroid deficiency (increasing lethargy, slow pulse, constipation, dry hair and skin), adrenal deficiency (weakness, orthostatic hypotension, hypoglycemia, fatigue, and weight loss), and gonadotropin deficiency (decreased libido, lethargy, and apathy).
Watch for anorexia in the patient with panhypopituitarism. Help plan a menu containing favorite foods—ideally, high-calorie foods. Monitor for weight loss or gain.
If the patient has trouble sleeping, encourage exercise during the day.
Record temperature, blood pressure, and heart rate every 4 to 8 hours. Check eyelids, nail beds, and skin for pallor, which indicates anemia.
Prevent infection by giving meticulous skin care. Because the patient’s skin is probably dry, use oil or lotion instead of soap. If body temperature is low, provide additional clothing and covers, as needed, to keep the patient warm.
Darken the room if the patient has a tumor that’s causing headaches and visual disturbances. Help with any activity that requires good vision such as reading the menu. The patient with bilateral hemianopia has impaired peripheral vision, so be sure to stand where he can see you, and advise the family to do the same.
During insulin testing, monitor closely for signs of hypoglycemia (initially, slow cerebration, tachycardia, diaphoresis, and nervousness, progressing to seizures). Keep dextrose 50% in water available for I.V. administration to correct hypoglycemia rapidly.
To prevent orthostatic hypotension, be sure to keep the patient supine during levodopa testing.
Instruct the patient to wear a medical identification bracelet. Teach him and his family members how to administer steroids rectally or parenterally in case of an emergency.
Refer the family of a child with dwarfism to the appropriate community resources for psychological counseling because the emotional stress caused by this disorder increases as the child becomes more aware of his condition.
Hyperpituitarism
Hyperpituitarism, also called acromegaly and gigantism, are chronic, progressive diseases marked by hormonal dysfunction and startling skeletal overgrowth. Although the prognosis depends on the causative factor, these disorders usually reduce life expectancy unless treated in a timely fashion.
Acromegaly occurs after epiphyseal closure, causing bone thickening and transverse bone growth and visceromegaly.
Gigantism begins before epiphyseal closure and causes proportional overgrowth of all body tissues. As the disease progresses, loss of other trophic hormones, such as thyroid-stimulating hormone, luteinizing hormone, follicle-stimulating hormone, and corticotropin, may cause dysfunction of the target organs.
CAUSES AND INCIDENCE
Typically, oversecretion of human growth hormone (hGH) produces changes throughout the body, resulting in acromegaly and, when oversecretion occurs before puberty, gigantism. Eosinophilic or mixed-cell adenomas of the anterior pituitary gland may cause this oversecretion but the etiology of the tumors themselves remains unclear. Occasionally, hGH levels are elevated in more than one family member, which suggests the possibility of a genetic cause.
The earliest clinical manifestations of acromegaly include soft-tissue swelling of the extremities and coarsening of facial features. This rare form of hyperpituitarism occurs equally among males and females, usually between ages 30 and 50. Annually, it affects 3 to 4 people per every million.
In gigantism, proportional overgrowth of all body tissues causes remarkable height increases of as much as 6” (15 cm) per year. Gigantism affects infants and children, causing them to attain as much as three times the normal height for their age. As adults, they may ultimately reach a height of more than 80” (203 cm). Gigantism is rare; there have only been 100 reported cases.
COMPLICATIONS
Increased hGH secretion—arthritis, carpal tunnel syndrome, osteoporosis, kyphosis, hypertension, arteriosclerosis, heart enlargement, heart failure
Acromegaly—blindness, neurologic disturbances
Acromegaly and gigantism—glucose intolerance, diabetes mellitus
SIGNS AND SYMPTOMS
Acromegaly develops slowly and typically produces diaphoresis, oily skin, hypermetabolism, and hypertrichosis. Severe headache, central nervous system impairment, bitemporal hemianopia, loss of visual acuity, and blindness may result from the intrasellar tumor compressing the optic chiasm or nerves.
Hypersecretion of hGH produces cartilaginous and connective tissue overgrowth, resulting in a characteristic hulking appearance, with an enlarged supraorbital ridge and thickened ears and nose. Prognathism, projection of the jaw, becomes marked and may interfere with chewing. Laryngeal hypertrophy, paranasal sinus enlargement, and thickening of the tongue cause the voice to sound deep and hollow. Distal phalanges display an arrowhead appearance on X-rays, and the fingers are thickened. Irritability, hostility, and various psychological disturbances may occur.
Prolonged effects of excessive hGH secretion include bowlegs, barrel chest, arthritis, osteoporosis, kyphosis, hypertension, and arteriosclerosis. Both gigantism and acromegaly may also cause signs of glucose intolerance and clinically apparent diabetes mellitus because of the insulin-antagonistic character of hGH. If acromegaly is left untreated, the patient is at risk for premature cardiovascular disease, colon polyps, and colon cancer.
Gigantism develops abruptly, producing some of the same skeletal abnormalities seen in acromegaly. As the disease progresses, the pituitary tumor enlarges and invades normal tissue, resulting in the loss of other trophic hormones, such as thyroid-stimulating hormone, luteinizing hormone, follicle-stimulating hormone, and corticotropin, thus causing the target organ to stop functioning.
DIAGNOSIS
Plasma hGH and insulin-like growth factor-1 (IGF-1) levels measured by radioimmunoassay typically are elevated. Because hGH secretion is pulsatile, the results of random sampling may be misleading. The glucose suppression test offers more reliable information. Glucose normally suppresses hGH secretion; therefore, a glucose infusion that doesn’t suppress the hormone level to below the accepted normal value of 5 nanograms (ng)/ml, when combined with characteristic clinical features, strongly suggests hyperpituitarism.
In addition, skull X-rays, computed tomography scan, arteriography, and magnetic resonance imaging determine the presence and extent of the pituitary lesion. Bone X-rays showing a thickening of the cranium (especially of frontal, occipital, and parietal bones) and of the long bones, as well as osteoarthritis in the spine, support this diagnosis.
TREATMENT
Treatment aims to curb overproduction of hGH through removal of the underlying tumor by cranial or transsphenoidal hypophysectomy or pituitary radiation therapy. In acromegaly, surgery is mandatory when a tumor causes blindness or other severe neurologic disturbances. Postoperative therapy often requires replacement of thyroid, cortisone, and gonadal hormones. Adjunctive treatment may include administration of bromocriptine or cabergoline and octreotide and postoperative conventional proton beam radiation, which inhibit hGH
synthesis. The therapeutic goal is to reach and maintain hGH levels less than 1 ng/ml and IGF-1 to normal range for age and gender, because at that level, life expectancy is restored to that of age-matched controls.
synthesis. The therapeutic goal is to reach and maintain hGH levels less than 1 ng/ml and IGF-1 to normal range for age and gender, because at that level, life expectancy is restored to that of age-matched controls.
SPECIAL CONSIDERATIONS
The extreme body changes characteristic of this disorder can cause severe psychological stress; therefore, emotional support to help the patient cope with his altered body image is an integral part of patient care.
Assess for skeletal manifestations, such as arthritis of the hands and osteoarthritis of the spine. Administer medications as ordered. To promote maximum joint mobility, perform or assist with range-of-motion exercises.
Evaluate muscle weakness, especially in the patient with late-stage acromegaly. Check the strength of his handclasp. If it’s very weak, help with tasks such as cutting food.
Keep the skin dry. Avoid using an oily lotion because the skin is already oily.
Test blood for glucose. Check for signs of hyperglycemia (fatigue, polyuria, and polydipsia).
Be aware that the tumor may cause visual problems. If the patient has hemianopia, stand where he can see you. Remember, this disease can also cause inexplicable mood changes. Reassure the family that these mood changes result from the disease and can be modified with treatment.
Before surgery, reinforce what the surgeon has told the patient, and try to allay the patient’s fear with a clear and honest explanation of the scheduled operation.
If the patient is a child, explain to his parents that such surgery prevents permanent soft-tissue deformities but won’t correct bone changes that have already taken place. Arrange for counseling, if necessary, to help the child and his parents cope with these permanent defects.
After surgery, diligently monitor vital signs and neurologic status. Immediately report increased urine output lasting longer than 2 hours, alteration in level of consciousness, unequal pupil size, changes in visual acuity, vomiting, falling pulse rate, or rising blood pressure. These changes may signal an increase in intracranial pressure due to intracranial bleeding or cerebral edema.
Check blood glucose level often. Remember, hGH levels usually fall rapidly after surgery, removing an insulin-antagonist effect in many patients and possibly precipitating hypoglycemia. Measure intake and output hourly, and report large increases. Transient diabetes insipidus, which sometimes occurs after surgery for hyperpituitarism, can cause such increases in urine output.
If the transsphenoidal approach is used, a large nasal packing is kept in place for several days. Because the patient must breathe through his mouth, give good mouth care. Pay special attention to the mucous membranes—which usually become dry—and the incision site under the upper lip, at the top of the gum line. The surgical site is packed with a piece of tissue generally taken from a midthigh donor site. Watch for cerebrospinal fluid leaks from the packed site, which may necessitate additional surgery to repair the leak. Look for increased external nasal drainage or drainage into the nasopharynx.
Encourage the patient to walk as soon as possible after surgery.
Before discharge, emphasize the importance of continuing hormone replacement therapy, if ordered. Make sure the patient and his family understand which hormones are to be taken and why as well as the correct times and dosages. Warn against stopping the hormones suddenly.
Advise the patient to wear a medical identification bracelet at all times and to bring his hormone replacement schedule with him whenever he returns to the health care facility.
Instruct the patient to have follow-up examinations for the rest of his life because a slight chance exists that the tumor that caused his condition may recur.
Diabetes insipidus
Diabetes insipidus (also called pituitary diabetes insipidus) is a disorder of water metabolism resulting from a deficiency of circulating vasopressin (also called antidiuretic hormone [ADH]). (See Mechanism of ADH deficiency, page 558.) It’s characterized by excessive fluid intake and hypotonic polyuria. The disorder may start in childhood or early adulthood (the median age of onset is 21) and is more common in males than in females. Incidence is slightly higher today than in the past. In uncomplicated diabetes insipidus, the prognosis is good with adequate water replacement and replacement of ADH by tablet or nasal spray, and patients usually lead normal lives.
CAUSES AND INCIDENCE
Diabetes insipidus results centrally from intracranial neoplastic or metastatic lesions,
hypophysectomy or other neurosurgery, a skull fracture, or head trauma that damages the neurohypophyseal structures. It can also result nephrogenically from infection, granulomatous disease, and vascular lesions; it may be idiopathic and, rarely, familial. (Note: Pituitary diabetes insipidus shouldn’t be confused with nephrogenic diabetes insipidus, a rare congenital disturbance of water metabolism that results from renal tubular resistance to vasopressin.)
hypophysectomy or other neurosurgery, a skull fracture, or head trauma that damages the neurohypophyseal structures. It can also result nephrogenically from infection, granulomatous disease, and vascular lesions; it may be idiopathic and, rarely, familial. (Note: Pituitary diabetes insipidus shouldn’t be confused with nephrogenic diabetes insipidus, a rare congenital disturbance of water metabolism that results from renal tubular resistance to vasopressin.)
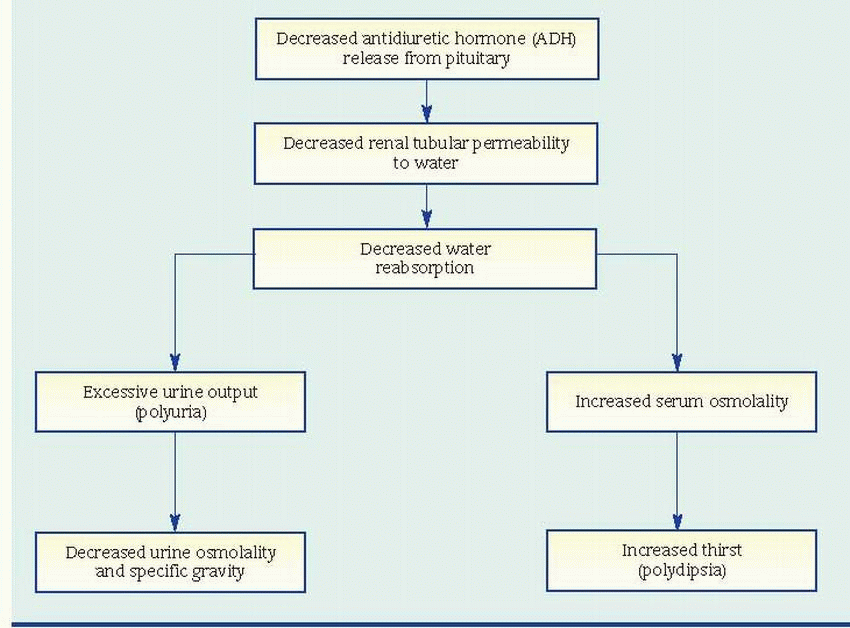
Normally, the hypothalamus synthesizes vasopressin. The posterior pituitary gland (or neurohypophysis) stores vasopressin and releases it into general circulation, where it causes the kidneys to reabsorb water by making the distal tubules and collecting duct cells water-permeable. The absence of vasopressin in diabetes insipidus allows the filtered water to be excreted in the urine instead of being reabsorbed.
Nephrogenic diabetes insipidus involves a defect in the parts of the kidneys that reabsorb water back into the bloodstream. It occurs less commonly than central diabetes insipidus. Nephrogenic diabetes insipidus may occur as an inherited disorder in which boys receive the abnormal gene that causes the disease on the X chromosome from their mothers. Nephrogenic diabetes insipidus may also be caused by diseases of the kidney (such as polycystic kidney disease) and the effects of certain drugs (such as lithium and amphotericin B) and as a result of hypercalcemia and hypokalemia.
Gestational diabetes insipidus occurs during pregnancy when an enzyme made by the placenta destroys antidiuretic hormone in the mother.
Diabetes insipidus is rare, affecting 1 in 25,000 people. Males and females are affected equally.
COMPLICATIONS
Hypovolemia
Hyperosmolality
Circulatory collapse
Loss of consciousness
Central nervous system damage
Laboratory values for patients with diabetes insipidus Diagnosis of diabetes insipidus requires evidence of vasopressin deficiency resulting in the kidneys’ inability to concentrate urine. Evidence of this can be seen in the patient’s laboratory values. | ||
Value | Normal | Diabetes insipidus (DI) |
Serum antidiuretic hormone | < 2.5 pg/ml | Decreased in central DI; may be normal with nephrogenic or psychogenic DI |
Serum osmolatity | 285 to 300 mOsm/kg | > 300 mOsm/kg |
Serum sodium | 136 to 145 mEq/L | >145 mEq/L |
Urine osmolality | 300 to 900 mOsm/kg | < 300 mOsm/kg |
Urine specific gravity | 1.005 to 1.030 | < 1.005 |
Urine output | 1 to 1.5 L/24 hr | 30 to 40 L/24 hr |
Fluid intake | 1 to 1.5 L/24 hr | > 50 L/24 hr |
SIGNS AND SYMPTOMS
The patient’s history typically shows an abrupt onset of extreme polyuria (usually 4 to 16 L/day of dilute urine but sometimes as much as 30 L/day). As a result, the patient is extremely thirsty and drinks great quantities of water to compensate for the body’s water loss. This disorder may also result in nocturia. In severe cases, it may lead to extreme fatigue from inadequate rest caused by frequent voiding and excessive thirst. Other characteristic features of diabetes insipidus include signs and symptoms of dehydration (poor tissue turgor, dry mucous membranes, constipation, muscle weakness, dizziness, and hypotension). These symptoms usually begin abruptly, commonly appearing within 1 to 2 days after a basal skull fracture, a stroke, or surgery. Relieving cerebral edema or increased intracranial pressure may cause all of these symptoms to subside just as rapidly as they began.
DIAGNOSIS
Urinalysis reveals almost colorless urine of low osmolality (50 to 200 mOsm/kg, less than that of plasma) and low specific gravity (<1.005). (See Laboratory values for patients with diabetes insipidus.)
Diagnosis requires evidence of vasopressin deficiency, resulting in the kidneys’ inability to concentrate urine during a water deprivation test.
In this test, after baseline vital signs, weight, and urine and plasma osmolalities are obtained, the patient is deprived of fluids and observed to make sure he doesn’t drink anything surreptitiously. Hourly measurements then record the total volume of urine output, body weight, urine osmolality or specific gravity, and plasma osmolality. Throughout the test, blood pressure and pulse rate must be monitored for signs of orthostatic hypotension. Fluid deprivation continues until the patient loses 3% of his body weight (indicating severe dehydration). When urine osmolality stops increasing in three consecutive hourly specimens, patients receive 5 units of aqueous vasopressin subcutaneously.
Hourly measurements of urine volume and specific gravity continue after subcutaneous injection of aqueous vasopressin. Patients with pituitary diabetes insipidus respond to exogenous vasopressin with decreased urine output and increased specific gravity. Patients with nephrogenic diabetes insipidus show no response to vasopressin.
TREATMENT
Mild cases require no treatment other than fluid intake to replace fluid lost. Until the cause of more severe cases of diabetes insipidus can be identified and eliminated, administration of various forms of vasopressin or of a vasopressin stimulant can control fluid balance and prevent dehydration. Vasopressin injection is an aqueous preparation that’s administered subcutaneously or I.M. several times a day because it’s effective for only 2 to 6 hours; this form of the drug is used in acute disease and as a diagnostic agent.
Desmopressin acetate (DDAVP) can be given by nasal spray that’s absorbed through the mucous membranes, or by injection given subcutaneously or I.V.; this drug is effective for 8 to 20 hours, depending on the dosage. It’s also available in tablet form, to be given at bedtime or in divided doses. Hydrochlorothiazide can be used in both central and nephrogenic diabetes insipidus. Indomethacin and amiloride are also used for nephrogenic diabetes insipidus. If nephrogenic diabetes insipidus is caused by medication (such as lithium [Eskalith]), stopping the medicine leads to kidney recovery.
SPECIAL CONSIDERATIONS
Patient care includes monitoring symptoms to ensure that fluid balance is restored and maintained.
Record fluid intake and output carefully. Maintain fluid intake that’s adequate to prevent severe dehydration. Watch for signs of hypovolemic shock, and monitor blood pressure and heart and respiratory rates regularly, especially during the water deprivation test. Check the patient’s weight daily.
If the patient is dizzy or has muscle weakness, keep the side rails up and assist him with walking.
Monitor urine specific gravity between doses. Watch for a decrease in specific gravity accompanied by increasing urine output, indicating the recurrence of polyuria and necessitating administration of the next dose of medication or a dosage increase.
Institute safety precautions for the patient who’s dizzy or who has muscle weakness.
If constipation develops, add more high-fiber foods and fruit juices to the patient’s diet. If necessary, obtain an order for a mild laxative such as milk of magnesia.
Provide meticulous skin and mouth care; apply petroleum jelly, as needed, to cracked or sore lips.
Before discharge, teach the patient how to monitor intake and output.
Instruct the patient to administer desmopressin by nasal spray only after the onset of polyuria—not before—to prevent excess fluid retention and water intoxication.
Tell the patient to report weight gain, which may indicate that his medication dosage is too high. Recurrence of polyuria, as reflected on the intake and output sheet, indicates that the dosage is too low.
Teach the parents of a child with diabetes insipidus about normal growth and development. Discuss how their child may differ from others at his developmental stage.
Encourage the parents to help identify the child’s strengths and to use them in developing coping strategies.
Refer the family for counseling if necessary.
Advise the patient with diabetes insipidus to wear a medical identification bracelet and to carry his medication with him at all times.
THYROID GLAND
Hypothyroidism in adults
Hypothyroidism, a state of low serum thyroid hormone, results from hypothalamic, pituitary, or thyroid insufficiency. The disorder can progress to life-threatening myxedema coma.
CAUSES AND INCIDENCE
Hypothyroidism results from inadequate production of thyroid hormone—usually because of dysfunction of the thyroid gland due to surgery (thyroidectomy), irradiation therapy (particularly with131I), inflammation, chronic autoimmune thyroiditis (Hashimoto’s disease) or, rarely, conditions such as amyloidosis and sarcoidosis. It may also result from pituitary failure to produce thyroid-stimulating hormone (TSH), hypothalamic failure to produce thyrotropin-releasing hormone, inborn errors of thyroid hormone synthesis, the inability to synthesize thyroid hormone because of iodine deficiency (usually dietary), or the use of antithyroid medications such as propylthiouracil. In patients with hypothyroidism, infection, exposure to cold, and sedatives may precipitate myxedema coma.
Hypothyroidism is more prevalent in females than males, and frequency increases with age; in the United States, incidence is rising significantly in people ages 40 to 50.
COMPLICATIONS
Cardiovascular—arteriosclerosis, ischemic heart disease, poor peripheral circulation, heart enlargement, heart failure, and pleural and pericardial effusion
Gastrointestinal—achlorhydria, pernicious anemia, megacolon, and intestinal obstruction
Bleeding tendencies
Iron deficiency anemia
Psychiatric disturbances
Carpal tunnel syndrome
Impaired fertility
Benign intracranial hypertension
Conductive or sensorineural deafness
Myxedema coma
SIGNS AND SYMPTOMS
Typically, the early clinical features of hypothyroidism are vague: fatigue, menstrual changes, hypercholesterolemia, forgetfulness, sensitivity to cold, unexplained weight gain, and constipation. As the disorder progresses, characteristic myxedematous signs and symptoms appear: decreasing mental stability; dry, flaky, inelastic skin; puffy face, hands, and feet; hoarseness; periorbital edema; upper eyelid droop; dry, sparse hair; and thick, brittle nails. (See Facial signs of myxedema.)
Cardiovascular involvement leads to decreased cardiac output, slow pulse rate, signs of poor peripheral circulation and, occasionally, an enlarged heart. Other common effects include anorexia, abdominal distention, menorrhagia, decreased libido, infertility, ataxia, intention tremor, and nystagmus. Reflexes show delayed relaxation time (especially in the Achilles tendon).
Progression to myxedema coma is usually gradual but when stress (such as hip fracture, infection, or myocardial infarction) aggravates severe or prolonged hypothyroidism, coma may develop abruptly. Clinical effects include progressive stupor, hypoventilation, hypoglycemia, hyponatremia, hypotension, and hypothermia.
DIAGNOSIS
Radioimmunoassay confirms hypothyroidism with low triiodothyronine (T3) and thyroxine (T4) levels. Supportive laboratory findings include:
increased TSH level when hypothyroidism is due to thyroid insufficiency; decreased TSH level when hypothyroidism is due to hypothalamic or pituitary insufficiency
elevated levels of serum cholesterol, alkaline phosphatase, and triglycerides
normocytic normochromic anemia
In myxedema coma, laboratory tests may also show low serum sodium levels, as well as decreased pH and increased partial pressure of carbon dioxide, indicating respiratory acidosis.
TREATMENT
Therapy for hypothyroidism consists of gradual thyroid replacement with levothyroxine (for low T4 levels) and, occasionally, liothyronine (for inadequate T3 levels).

During myxedema coma, effective treatment supports vital functions while restoring euthyroidism. To support blood pressure and pulse rate, treatment includes I.V. administration of levothyroxine and hydrocortisone to correct possible pituitary or adrenal insufficiency. Hypoventilation requires oxygenation and respiratory support. Other supportive measures include fluid replacement and antibiotics for infection.
SPECIAL CONSIDERATIONS
To manage the hypothyroid patient:
Provide a high-bulk, low-calorie diet and encourage activity to combat constipation and promote weight loss. Administer cathartics and stool softeners, as needed.
After thyroid replacement therapy begins, watch for symptoms of hyperthyroidism, such as restlessness, sweating, and excessive weight loss.
Tell the patient to report any signs of aggravated cardiovascular disease, such as chest pain and tachycardia.
To prevent myxedema coma, tell the patient to continue his course of thyroid medication even if his symptoms subside.
Warn the patient to report infection immediately and to make sure any physician who prescribes drugs for him knows about the underlying hypothyroidism.
Treatment of myxedema coma requires supportive care:
Check frequently for signs of decreasing cardiac output such as falling urine output.
Monitor temperature until stable. Provide extra blankets and clothing and a warm room to compensate for hypothermia. Rapid rewarming may cause vasodilation and vascular collapse.
Record intake and output and daily weight. As treatment begins, urine output should increase and body weight decrease; if not, report this immediately.
Turn the edematous bedridden patient every 2 hours, and provide skin care, particularly around bony prominences.
Avoid sedation when possible or reduce dosage because hypothyroidism delays metabolism of many drugs.
Maintain a patent I.V. line. Monitor serum electrolyte levels carefully when administering I.V. fluids.
Monitor vital signs carefully when administering levothyroxine because rapid correction of hypothyroidism can cause adverse cardiac effects. Report chest pain or tachycardia immediately. Watch for hypertension and heart failure in the elderly patient.
Check arterial blood gas values for hypercapnia and hypoxia to determine whether the patient who’s severely myxedematous requires ventilatory assistance.
Because myxedema coma may have been precipitated by an infection, check possible sources of infection, such as blood or urine, and obtain sputum cultures.
Hypothyroidism in children
Deficiency of thyroid hormone secretion during fetal development or early infancy results in infantile cretinism (congenital or neonatal hypothyroidism). Untreated hypothyroidism is characterized in infants by respiratory difficulties, persistent jaundice, and hoarse crying; in older children, by stunted growth (dwarfism), bone and muscle dystrophy, and mental deficiency.
Cretinism is three times more common in females than in males. Early diagnosis and treatment allow the best prognosis; infants treated before age 3 months usually grow and develop normally. However, athyroidic children who remain untreated beyond age 3 months and children with acquired hypothyroidism who remain untreated beyond age 2 suffer irreversible mental retardation; their skeletal abnormalities are reversible with treatment.
CAUSES AND INCIDENCE
In infants, cretinism usually results from defective embryonic development that causes congenital absence or underdevelopment of the thyroid gland. The next most common cause can be traced to an inherited enzymatic defect in the synthesis of thyroxine (T4) caused by an autosomal recessive gene. Less frequently, antithyroid drugs taken during pregnancy produce cretinism in infants. In children older than age 2, cretinism usually results from chronic autoimmune thyroiditis.
COMPLICATIONS
Severe mental retardation
Skeletal malformations (dwarfism, epiphyseal degeneration, and bone and muscle dystrophy)
SIGNS AND SYMPTOMS
The weight and length of an infant with infantile cretinism appear normal at birth, but characteristic signs of hypothyroidism develop by the time he’s 3 to 6 months old. In a breastfed infant the onset of most symptoms may be delayed until weaning because breast milk contains small amounts of thyroid hormone.
Typically, an infant with cretinism sleeps excessively, seldom cries (except for occasional hoarse crying), and is inactive. Because of this, his parents may describe him as a “good baby— no trouble at all.” However, such behavior actually results from lowered metabolism and progressive mental impairment. The infant with cretinism also exhibits abnormal deep tendon reflexes, hypotonic abdominal muscles, a protruding abdomen, and slow, awkward movements. He has feeding difficulties, develops constipation and, because his immature liver can’t conjugate bilirubin, becomes jaundiced.
His large, protruding tongue obstructs respiration, making breathing loud and noisy and forcing him to open his mouth to breathe. He may have dyspnea on exertion, anemia, abnormal facial features—such as a short forehead; puffy, wide-set eyes (periorbital edema); wrinkled eyelids; and a broad, short, upturned nose—and a dull expression, resulting from mental retardation. His skin is cold and mottled because of poor circulation, and his hair is dry, brittle, and dull. Teeth erupt late and tend to decay early; body temperature is below normal; and pulse rate is slow.
In the child who acquires hypothyroidism after age 2, appropriate treatment can prevent mental retardation. However, growth retardation becomes apparent in short stature (due to delayed epiphyseal maturation, particularly
in the legs), obesity, and a head that appears abnormally large because the arms and legs are stunted. An older child may show delayed or accelerated sexual development.
in the legs), obesity, and a head that appears abnormally large because the arms and legs are stunted. An older child may show delayed or accelerated sexual development.
DIAGNOSIS
A high serum level of thyroid-stimulating hormone (TSH), associated with low triiodothyronine and T4 levels, points to cretinism. Because early detection and treatment can minimize the effects of cretinism, many states require measurement of infant thyroid hormone levels at birth.
Thyroid scan and radioactive iodine uptake tests show decreased uptake levels and confirm the absence of thyroid tissue in athyroidic children. Increased gonadotropin levels are compatible with sexual precocity in older children and may coexist with hypothyroidism.
Electrocardiogram shows bradycardia and flat or inverted T waves in untreated infants. Hip, knee, and thigh X-rays reveal absence of the femoral or tibial epiphyseal line and delayed skeletal development that’s markedly inappropriate for the child’s chronological age. A low T4 level associated with a normal TSH level suggests hypothyroidism secondary to hypothalamic or pituitary disease, a rare condition.
TREATMENT
Early detection is mandatory to prevent irreversible mental retardation and permit normal physical development. Treatment of infants younger than age 1 consists of replacement therapy with oral levothyroxine, beginning with moderate doses. Dosage gradually increases to levels sufficient for lifelong maintenance. (Rapid increase in dosage may precipitate thyrotoxicity.) Doses are proportionately higher in children than in adults because children metabolize thyroid hormone more quickly. Therapy in older children includes levothyroxine.
SPECIAL CONSIDERATIONS
Prevention, early detection, comprehensive parent teaching, and psychological support are essential. Know the early signs. Be especially wary if parents emphasize how good and how quiet their new baby is.
During early management of infantile cretinism, monitor blood pressure and pulse rate; report hypertension and tachycardia immediately. But remember—normal infant heart rate is approximately 120 beats per minute. If the infant’s tongue is unusually large, position him on his side and observe him frequently to prevent airway obstruction. Check rectal temperature every 2 to 4 hours. Keep the infant warm and his skin moist.
Inform parents that the child will require lifelong treatment with thyroid supplements. Teach them to recognize signs of overdose: rapid pulse rate, irritability, insomnia, fever, sweating, and weight loss. Stress the need to comply with the treatment regimen to prevent further mental impairment.
Provide support to help parents deal with a child who may be mentally retarded. Help them adopt a positive but realistic attitude, and focus on their child’s strengths rather than his weaknesses. Encourage them to provide stimulating activities to help the child reach his maximum potential. Refer them to the appropriate community resources for support.
To prevent infantile cretinism, emphasize the importance of adequate nutrition during pregnancy, including iodine-rich foods and the use of iodized salt or, in case of sodium restriction, an iodine supplement.
Thyroiditis
Inflammation of the thyroid gland occurs as autoimmune thyroiditis (long-term inflammatory disease), subacute granulomatous thyroiditis (self-limiting inflammation), Riedel’s thyroiditis (rare, invasive fibrotic process), and miscellaneous thyroiditis (acute suppurative, chronic infective, and chronic noninfective forms).
CAUSES AND INCIDENCE
Autoimmune thyroiditis is due to anti-thyroid antibodies in the blood. It may cause inflammation and lymphocytic infiltration (Hashimoto’s thyroiditis). Glandular atrophy (myxedema) and Graves’ disease are linked to autoimmune thyroiditis.
Subacute granulomatous thyroiditis usually follows mumps, influenza, coxsackievirus, or adenovirus infection. Riedel’s thyroiditis is a rare condition of unknown etiology.
Miscellaneous thyroiditis results from bacterial invasion of the gland in acute suppurative thyroiditis; tuberculosis, syphilis, actinomycosis, or other infectious agents in the chronic infective form; and sarcoidosis and amyloidosis in chronic noninfective thyroiditis. Postpartum thyroiditis (silent thyroiditis) is another autoimmune disorder associated with transient thyroiditis in females within 1 year after delivery.
COMPLICATIONS
Hasmimoto’s—compression of surrounding tissues
Subacute—permanent hypothyroid or hyperthyroid condition
Pyrogenic—rupture of abscess into mediastinum, trachea, or esophagus
Riedel’s—hypothyroidism, tracheal or esophageal compression, and necrosis or hemorrhage of compressed tissue
SIGNS AND SYMPTOMS
Autoimmune thyroiditis is usually asymptomatic and commonly occurs in females, with peak incidence in middle age. It’s the most prevalent cause of spontaneous hypothyroidism.
In subacute granulomatous thyroiditis, moderate thyroid enlargement may follow an upper respiratory tract infection or a sore throat. The thyroid may be painful and tender, and dysphagia may occur.
In Riedel’s thyroiditis, the gland enlarges slowly as it’s replaced by hard, fibrous tissues. This fibrosis may compress the trachea or the esophagus. The thyroid feels firm.
Clinical effects of miscellaneous thyroiditis are characteristic of pyogenic infection: fever, pain, tenderness, and reddened skin over the gland.
DIAGNOSIS
Precise diagnosis depends on the type of thyroiditis:
Autoimmune: high titers of thyroglobulin and microsomal antibodies present in serum
Subacute granulomatous: elevated erythrocyte sedimentation rate, increased thyroid hormone levels, decreased thyroidal radioiodine uptake
Chronic infective and noninfective: varied findings, depending on underlying infection or other disease
TREATMENT
Appropriate treatment varies with the type of thyroiditis. Drug therapy includes levothyroxine for accompanying hypothyroidism, analgesics and anti-inflammatory drugs for mild subacute granulomatous thyroiditis, propranolol for transient hyperthyroidism, and steroids for severe episodes of acute inflammation. Suppurative thyroiditis requires antibiotic therapy. A partial thyroidectomy may be necessary to relieve tracheal or esophageal compression in Riedel’s thyroiditis.
SPECIAL CONSIDERATIONS
Before treatment, obtain a patient history to identify underlying diseases that may cause thyroiditis, such as tuberculosis or a recent viral infection.
Check the patient’s vital signs, and examine her neck for unusual swelling, enlargement, or redness. Provide a liquid diet if she has difficulty swallowing, especially when due to fibrosis. If the neck is swollen, measure and record the circumference daily to monitor progressive enlargement.
Administer antibiotics as indicated, and report and record elevations in temperature.
Instruct the patient to watch for and report signs of hypothyroidism (lethargy, restlessness, sensitivity to cold, forgetfulness, and dry skin), especially if she has Hashimoto’s thyroiditis, which often causes hypothyroidism.
Check for signs of hyperthyroidism (nervousness, tachycardia, tremor, and weakness), which commonly occurs in subacute thyroiditis.
After thyroidectomy, check vital signs every 15 to 30 minutes until the patient’s condition stabilizes. Stay alert for signs of tetany secondary to accidental parathyroid injury during surgery. Keep 10% calcium gluconate available for I.V. use if needed. Assess dressings frequently for excessive bleeding. Watch for signs of airway obstruction, such as difficulty in talking or increased swallowing; keep tracheotomy equipment handy.
Explain to the patient that she’ll need lifelong thyroid hormone replacement therapy if hypothyroidism occurs. Tell her to watch for signs of overdosage, such as nervousness and palpitations.
Simple goiter
Simple (or nontoxic) goiter is a thyroid gland enlargement that isn’t caused by inflammation or a neoplasm, and is commonly classified as endemic or sporadic. Endemic goiter usually results from inadequate dietary intake of iodine associated with such factors as iodine-depleted soil or malnutrition. Sporadic goiter follows ingestion of certain drugs or foods.
Simple goiter affects more females than males, especially during adolescence, pregnancy, and menopause, when the body’s demand for thyroid hormone increases. Sporadic goiter affects no particular population segment. With appropriate treatment, the prognosis is good for either type of goiter.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


