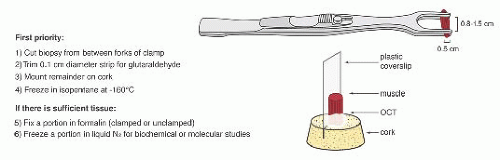
 FIGURE 4.1 Muscle biopsy in a clamp with suggested priorities for processing a routine skeletal muscle biopsy specimen. OCT, optimal cutting temperature embedding medium. |
When the pH of the incubating solution is reduced to the acidic range (pH 4.2) in the so-called reverse ATPase reaction, type 1 fibers are very dark and type 2 fibers are very light. At a slightly less acidic pH 4.6, type 2A fibers are very light and the staining intensity of type 2B fibers is intermediate. Commercial antibodies to slow (type 1) and fast (type 2) myosin are now available. By means of automated immunoperoxidase staining, fiber typing with results similar to those obtained with ATPase reactions can be achieved in either cryosections or paraffin sections (3,17).
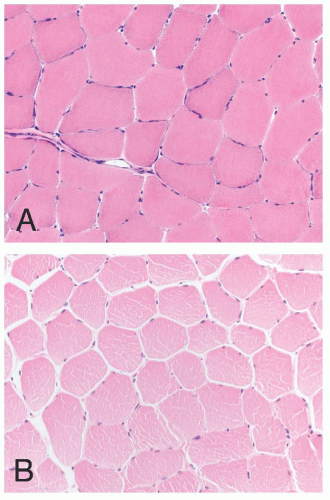 FIGURE 4.2 Normal muscle in transverse section (A, frozen; B, paraffin embedded). The fibers are typically polygonal, and the nuclei are located peripherally. Empty space between muscle fibers is a shrinkage artifact, which is typically more pronounced in paraffin sections (H&E). |
TABLE 4.1 Fiber Typing in Skeletal Muscle | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||
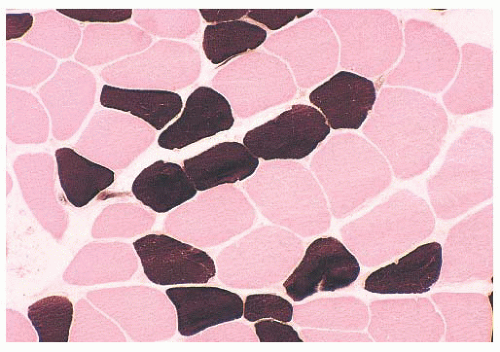 FIGURE 4.3 Fiber types in normal muscle. In the alkaline adenosine triphosphatase (ATPase) reaction, type 1 fibers are light and type 2 fibers are dark (ATPase at pH 9.4 and counterstained with eosin). |
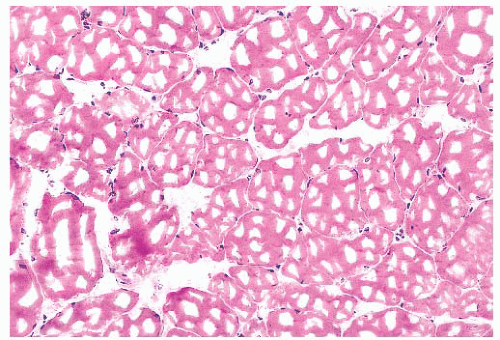 FIGURE 4.4 Freezing artifact. Extensive vacuolar change can be caused by the formation of ice crystals in the muscle during improper freezing. Vacuoles can be a variety of geometric shapes (H&E). |
plastic disposable clamps that only partially prevent contraction are used or when the muscle is injected with local anesthetic. Contraction artifact renders muscle particularly unfit for electron microscopy. The orderly structural landmarks are obliterated by myofibrillar fragmentation and disorientation.
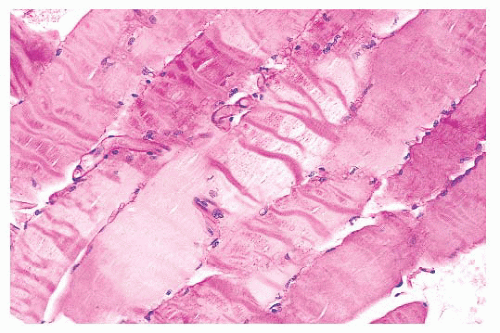 FIGURE 4.5 Contraction artifact. Darker contraction bands and disrupted lucent zones are seen in several longitudinally oriented fibers (PAS). |
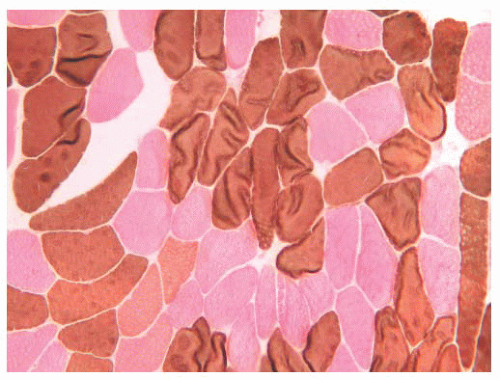 FIGURE 4.6 Muscle fiber wrinkling. Frozen sections may partially lift off the slide during staining, creating artifacts such as stripes and ring structures in the fibers (ATPase, pH 9.4, counterstained with eosin). |
TABLE 4.2 Diseases with Prominent Type 1 Fiber Atrophy/Smallness | ||||
|---|---|---|---|---|
|
in myasthenia gravis, acute denervation, disuse, and systemic malignancy (20). In our experience, more than half of such cases are attributable to corticosteroid therapy (Table 4.3). Hypertrophy restricted to type 1 fibers is relatively specific for infantile spinal muscular atrophy (SMAI), although such hypertrophy may develop in normal athletes who undergo endurance training. Type 2 fiber hypertrophy has been reported in runners, notably sprinters, and in congenital fibertype disproportion. Non-fiber type-selective alterations in fiber size are somewhat uninformative from a diagnostic standpoint. Denervation accounts for a large proportion of cases of nonselective atrophy but certainly not all of them. Hypertrophy involving both major fiber types is frequently noted in muscular dystrophy, inclusion body myositis, myotonia congenita, and acromegaly.
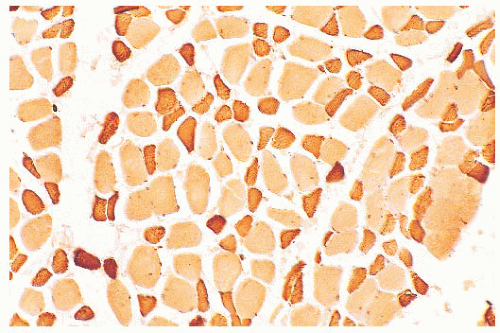 FIGURE 4.7 Type 2 fiber atrophy in a patient with malignancy and cachexia (immunostain for fast myosin). |
TABLE 4.3 Diseases with Prominent Type 2 Fiber Atrophy | |
|---|---|
|
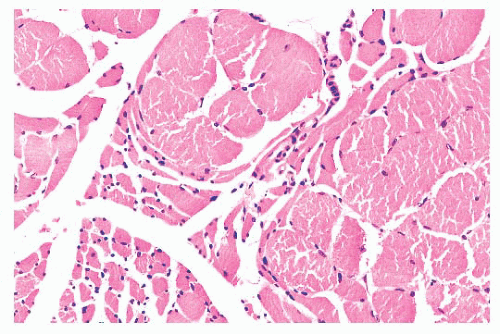 FIGURE 4.8 Chronic neurogenic atrophy. Angulated atrophic fibers arranged in groups are denervated motor units. The large size of the groups is suggestive of a chronic neuropathic process. |
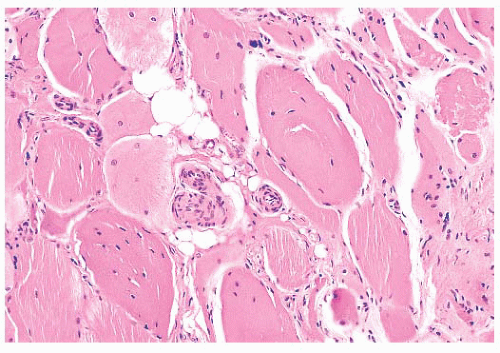 FIGURE 4.9 Internally placed nuclei. Many fibers contain one or more internal nuclei (H&E). |
each space, the extensions of the plasma membrane remain continuous around the dividing portions of the cell. Fiber splitting is typically more conspicuous in chronic necrotizing myopathies (e.g., muscular dystrophies or inflammatory myopathies) (21). Internalization of capillaries sometimes accompanies the fiber splitting process (see panel A of Fig. 4.36).
TABLE 4.4 Conditions in Which Internal Nuclei Have Diagnostic Significance | |
|---|---|
|
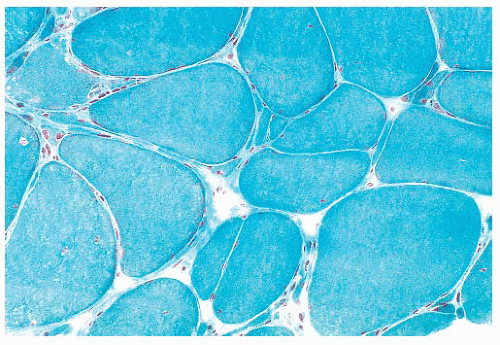 FIGURE 4.10 Fiber splitting. Several hypertrophic fibers are seen. The fiber at the bottom and center has been split into two smaller subunits (Gomori trichrome). |
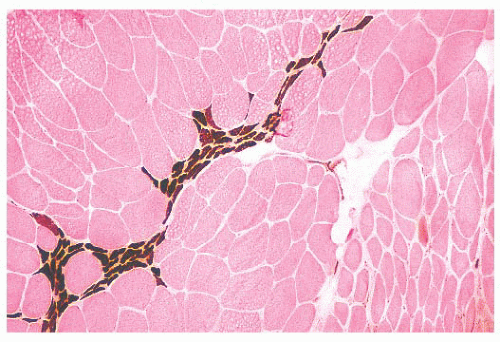 FIGURE 4.11 Fiber type grouping in denervation with reinnervation. Grouping of fiber types replaces the normal checkerboard staining pattern (ATPase at pH 9.4). |
TABLE 4.5 Inflammation Seen in the Biopsy Specimen | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
In systemic lupus erythematosus, the affected vessels are smaller in caliber, and they exhibit fibrinoid necrosis. Areas of vascular injury attract neutrophils, and the remnant of their breakdown, nuclear dust, is also seen. With the use of immunohistochemical techniques, deposits of immunoglobulin and complement can be demonstrated at sites of vascular injury in both conditions.
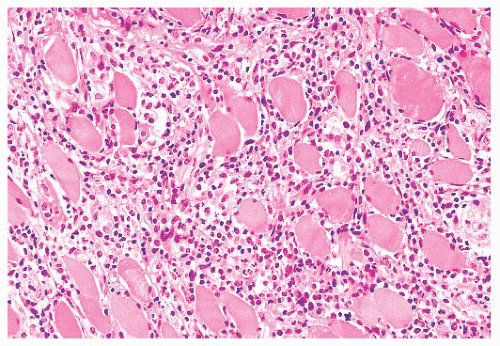 FIGURE 4.12 Inflammatory myopathy. Sheets of lymphocytes surround muscle fibers and expand the endomysium (H&E). |
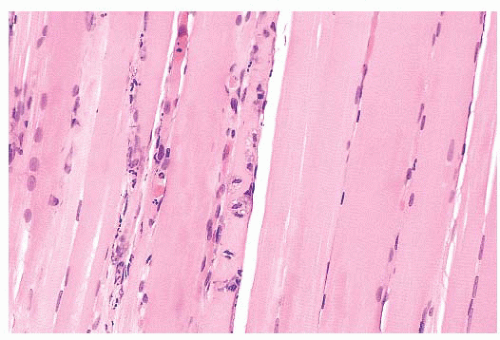 FIGURE 4.13 Fiber necrosis (myonecrosis). The necrotic process in the fiber at the center of this longitudinal section is recognized by a loss of cross-striations and early phagocytosis (H&E). |
TABLE 4.6 Fiber Necrosis Seen in the Biopsy Specimen | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
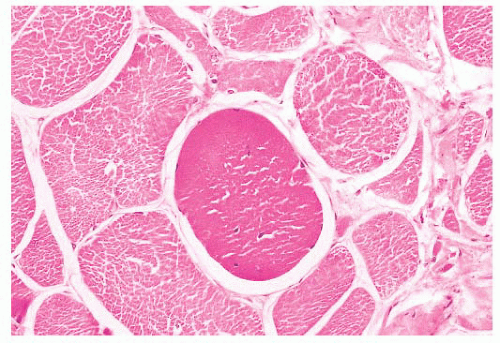 FIGURE 4.14 Hyaline fiber. The fiber in the center of the photograph is rounded, and it has dark, opaque sarcoplasm (H&E). |
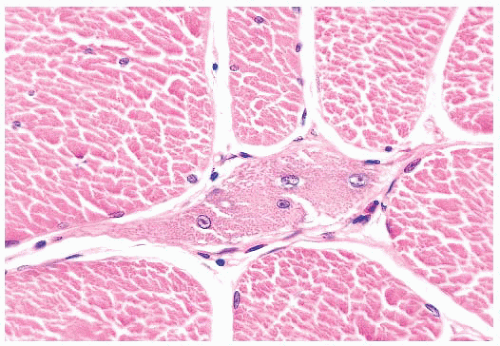 FIGURE 4.15 Regeneration. The nuclei of regenerating muscle fibers are large and vesicular with prominent nucleoli. The sarcoplasm is basophilic (H&E). |
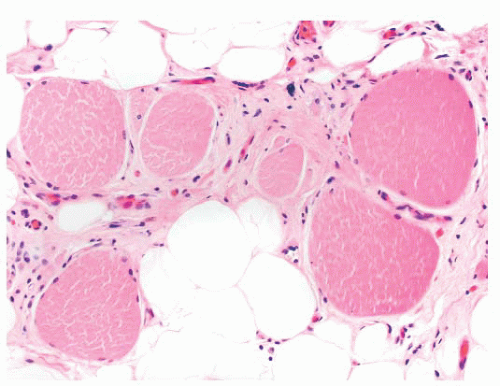 FIGURE 4.16 End-stage muscle. Atrophy, hypertrophy, pyknotic nuclear clusters, endomysial fibrosis, and fatty replacement are common features of end-stage muscle, regardless of the underlying neuromuscular disorder. |
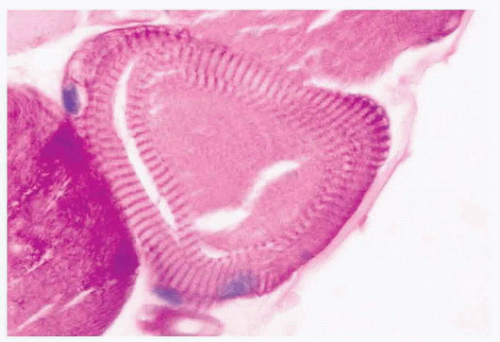 FIGURE 4.17 Ring fiber. Circumferential orientation of the peripheral myofibrils produces a striated ring that encircles a transversely sectioned muscle fiber (PAS). |
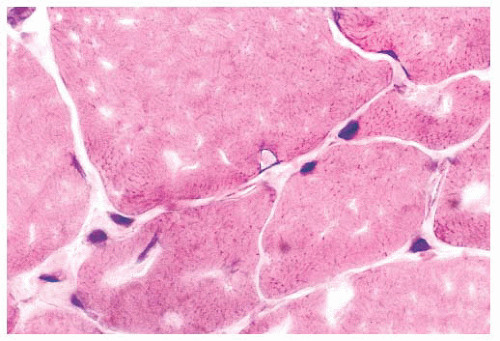 FIGURE 4.18 Intranuclear inclusion. An intranuclear inclusion is shown at the center of the photomicrograph. The inclusion is eosinophilic and smudged; it is located within a muscle fiber nucleus. The biopsy is from a patient with inclusion body myositis (H&E). |
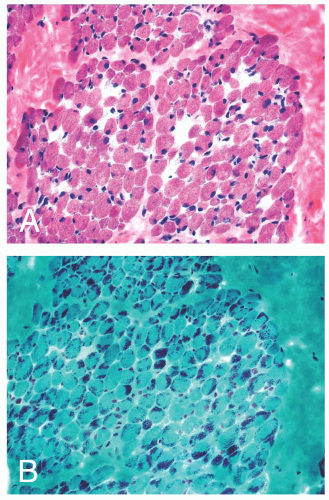 FIGURE 4.19 Nemaline myopathy. Nonspecific myopathic features of muscle fiber atrophy and hypertrophy are typically the predominant pathology in H&E stained sections (A). Collections of dark, granular to rod-shaped inclusions are striking after staining with Gomori trichrome (B). |
depleted or absent mitochondria (Fig. 4.23). Large cores are often also evident with H&E and Gomori trichrome stains. Ultrastructurally and in resin sections, distinguishing between structured and unstructured cores is possible (30). The crossbanding pattern is evident in the structured core, whereas cross-striations are absent from the unstructured core, which perhaps represents a later stage in core development. Cores cannot be regarded as a specific pathologic finding because they occur in a variety of diseases. However, the muscle fibers of several inherited myopathies contain cores, and one nosologic group, the core myopathies, is defined by them. As a nonspecific change, cores are present in less than 10% of fibers, whereas they are numerous and are located more often in type 1 fibers in central core disease. Although cores are single and centrally placed within the fiber in classic central core disease, they may be multiple and eccentric in other conditions.
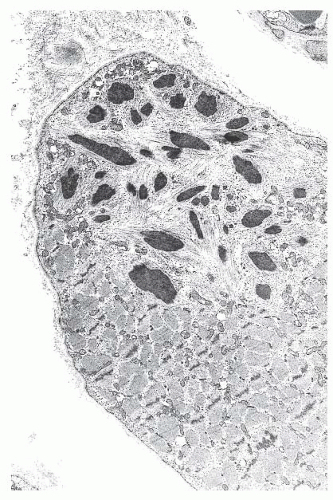 FIGURE 4.20 Nemaline myopathy ultrastructure. Nemaline rods are osmiophilic, resembling the electron density of Z-bands (electron micrograph). |
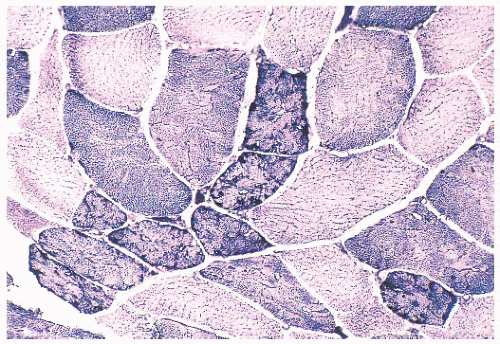 FIGURE 4.21 Mottled fibers. The sarcoplasm appears moth eaten as a result of the presence of patchy areas of poor staining (NADH). |
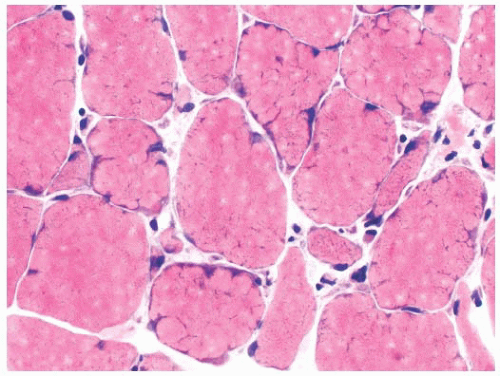 FIGURE 4.22 Lobulated fibers. Several muscle fibers in this image are divided into lobules by septa occupied by mitochondria (H&E). |
outer normal portion of the fiber. The term targetoid refers to target-like, sharply demarcated regions that lack the intermediate zone or rim of increased oxidative enzyme activity. Targetoid change and cores essentially are morphologically identical, but the term targetoid is typically used in neuropathic disease and cores in myopathic disease.
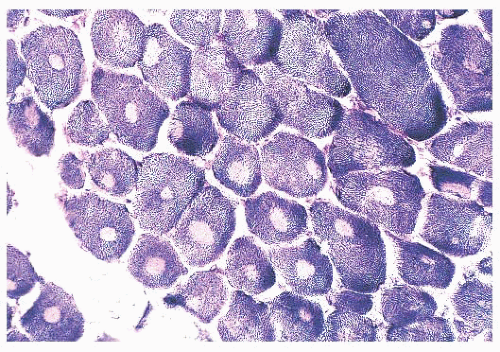 FIGURE 4.23 Cores. Muscle fibers contain cores (sharply demarcated areas of reduced enzyme activity) in several types of inherited myopathies. In this patient with central core myopathy, single cores are present within many muscle fibers (NADH). |
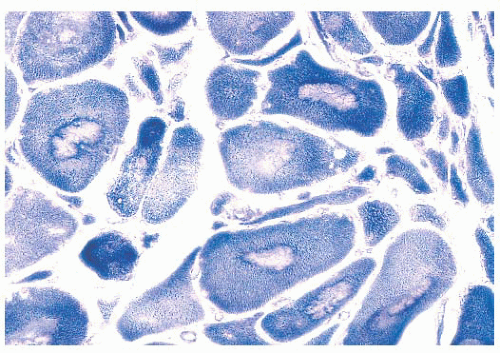 FIGURE 4.24 Target fibers. In target fibers, an inner, unstained zone is surrounded by a rim of increased enzyme activity (NADH). Targets are seen in the context of neurogenic atrophy, and angulated atrophic fibers are present in this image. |
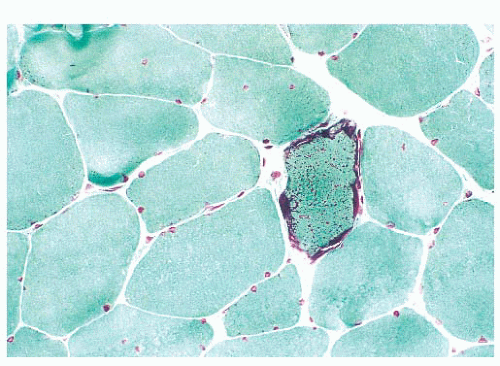 FIGURE 4.25 Ragged red fiber. Collections of mitochondria appear as red-stained, irregular, subsarcolemmal areas within the involved fiber (Gomori trichrome). |
and visible primarily in PAS stains, they are probably of no clinical significance.
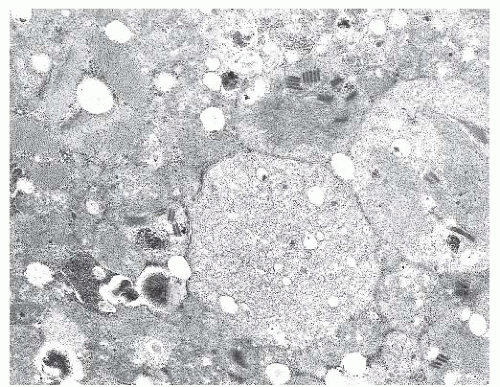 FIGURE 4.26 Mitochondrial myopathy. Ultrastructurally, a ragged red fiber shows numerous mitochondria. Here, some of the mitochondria are abnormally large, and some contain paracrystalline inclusions (electron micrograph). |
TABLE 4.7 Sarcoplasmic Vacuoles Seen in the Biopsy Specimen | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
from the autophagic vacuole, which classically is membrane bound and is a repository for the debris of autodigestion. In at least two genetic diseases, autophagic vacuoles are numerous (39,40). These vacuoles are rimmed by membrane that expresses proteins typically found in the sarcolemma. Enzyme histochemistry or immunostaining for these sarcolemmal proteins demonstrate strikingly numerous cytoplasmic vacuoles in Danon disease and X-linked myopathy with excessive autophagy (XMEA) (see Fig. 4.36).
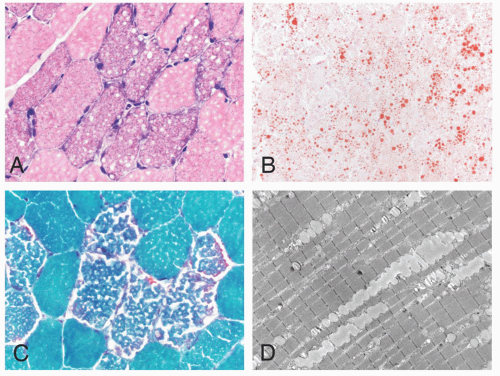 FIGURE 4.27 Lipid storage myopathy. Numerous vacuoles are evident in the sarcoplasm of affected muscle fibers (A, H&E; C, Gomori trichrome). The vacuoles contain neutral lipid that can be demonstrated by histochemistry (B, oil red O) or by electron microscopy (D). |
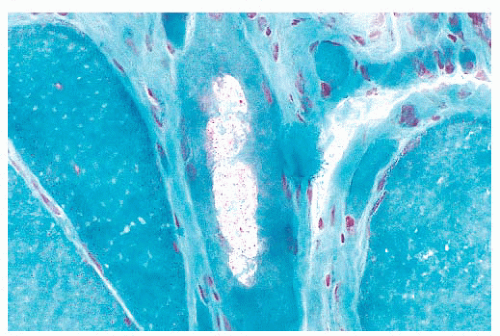 FIGURE 4.28 Rimmed vacuole. Aggregates of lysosomes have the appearance of rimmed vacuoles in frozen sections. The muscle fiber in the center contains rimmed vacuoles with red, granular material (Gomori trichrome). |
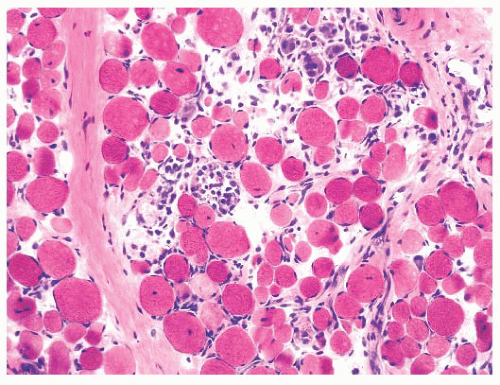 FIGURE 4.29 Muscular dystrophy histopathology. The classic histopathologic features of muscular dystrophies are illustrated here: myonecrosis and regeneration without significant lymphocytic inflammation, internally placed nuclei, muscle fiber atrophy or hypertrophy, and endomysial fibrosis (H&E). |
TABLE 4.8 Inherited Myopathiesa | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


