Multiple Myeloma
KEY CONCEPTS
![]() Multiple myeloma (MM) is a cancer that develops in plasma cells, leading to excessive production of a monoclonal immunoglobulin.
Multiple myeloma (MM) is a cancer that develops in plasma cells, leading to excessive production of a monoclonal immunoglobulin.
![]() Most patients have skeletal involvement at the time of diagnosis with associated bone pain and fractures. Anemia, hypercalcemia, and renal failure may also be present. A bone marrow biopsy with 10% or more plasma cells and an M-protein spike on plasma or urine electrophoresis confirms the diagnosis.
Most patients have skeletal involvement at the time of diagnosis with associated bone pain and fractures. Anemia, hypercalcemia, and renal failure may also be present. A bone marrow biopsy with 10% or more plasma cells and an M-protein spike on plasma or urine electrophoresis confirms the diagnosis.
![]() Most patients require treatment after diagnosis, but treatment can be deferred in patients with smoldering (asymptomatic) MM. In patients with symptomatic disease, treatment produces benefits in various measures of survival and quality of life.
Most patients require treatment after diagnosis, but treatment can be deferred in patients with smoldering (asymptomatic) MM. In patients with symptomatic disease, treatment produces benefits in various measures of survival and quality of life.
![]() Thalidomide, lenalidomide, or bortezomib plus dexamethasone are commonly used induction regimens. They produce higher complete remission rates compared with the classic regimens of melphalan plus prednisone and VAD (vincristine, doxorubicin, and dexamethasone). The increased response rate is at the expense of significant grade III and IV toxicity, which can include myelosuppression, venous thromboembolism (VTE), and neuropathy depending on the regimen used. These novel agents can be added to chemotherapy (melphalan, liposomal doxorubicin, cyclophosphamide, or VAD-like chemotherapy) as part of induction and results in substantially higher response rates. Novel agents can also be combined to produce more active regimens.
Thalidomide, lenalidomide, or bortezomib plus dexamethasone are commonly used induction regimens. They produce higher complete remission rates compared with the classic regimens of melphalan plus prednisone and VAD (vincristine, doxorubicin, and dexamethasone). The increased response rate is at the expense of significant grade III and IV toxicity, which can include myelosuppression, venous thromboembolism (VTE), and neuropathy depending on the regimen used. These novel agents can be added to chemotherapy (melphalan, liposomal doxorubicin, cyclophosphamide, or VAD-like chemotherapy) as part of induction and results in substantially higher response rates. Novel agents can also be combined to produce more active regimens.
![]() Bortezomib-based regimens are commonly used to treat newly diagnosed patients with high-risk disease and patients with relapsed MM.
Bortezomib-based regimens are commonly used to treat newly diagnosed patients with high-risk disease and patients with relapsed MM.
![]() Lenalidomide is more potent and better tolerated than thalidomide and is the most commonly used immunomodulatory drug.
Lenalidomide is more potent and better tolerated than thalidomide and is the most commonly used immunomodulatory drug.
![]() A host of new drugs are being studied and integrated into treatment of relapsed MM, including carfilzomib, pomalidomide, vorinostat, and bendamustine. Carfilzomib is a very active agent and is currently being studied as induction therapy in newly diagnosed patients.
A host of new drugs are being studied and integrated into treatment of relapsed MM, including carfilzomib, pomalidomide, vorinostat, and bendamustine. Carfilzomib is a very active agent and is currently being studied as induction therapy in newly diagnosed patients.
![]() Melphalan plus prednisone is not used in transplant candidates as part of induction but commonly used in transplant-ineligible patients combined with thalidomide, lenalidomide, or bortezomib.
Melphalan plus prednisone is not used in transplant candidates as part of induction but commonly used in transplant-ineligible patients combined with thalidomide, lenalidomide, or bortezomib.
![]() Autologous hematopoietic stem cell transplantation (HSCT) is used after induction in patients with reasonably good performance status to maximize complete remissions and prolong survival. Combining autologous HSCT with allogeneic HSCT must be considered investigational and should be performed under clinical trial.
Autologous hematopoietic stem cell transplantation (HSCT) is used after induction in patients with reasonably good performance status to maximize complete remissions and prolong survival. Combining autologous HSCT with allogeneic HSCT must be considered investigational and should be performed under clinical trial.
![]() Maintenance therapies can be used in both transplant-eligible and -ineligible patients. Current regimens usually include lenalidomide or bortezomib with the intent of increasing response rates and progression-free survival.
Maintenance therapies can be used in both transplant-eligible and -ineligible patients. Current regimens usually include lenalidomide or bortezomib with the intent of increasing response rates and progression-free survival.
![]() Bisphosphonates are used to treat bone disease associated with MM, which results in decreased pain and skeletal-related events and improvement in quality of life.
Bisphosphonates are used to treat bone disease associated with MM, which results in decreased pain and skeletal-related events and improvement in quality of life.
![]() Salvage therapy for patients with relapsed or refractory MM can include any of the prior listed therapies, depending on performance status of the patient, risk category of the patient, and prior treatments used for induction.
Salvage therapy for patients with relapsed or refractory MM can include any of the prior listed therapies, depending on performance status of the patient, risk category of the patient, and prior treatments used for induction.
INTRODUCTION
Multiple myeloma (MM) is a genetically complex and an increasingly more common hematologic malignancy that develops in plasma cells or immunoglobulin-producing B lymphocytes.1,2 The plasma cells produce excessive monoclonal immunoglobulins that can be measured in the plasma or urine. As a result of the various bone-mobilizing cytokines secreted from the MM clone and bone marrow stromal cells, patients often have skeletal involvement at diagnosis. MM is often sensitive to chemotherapy initially, but drug resistance develops relatively rapidly. Although therapy is not currently curative, MM has been a remarkable example of bench-to-bedside translation in new drug development. In particular, the proteasome inhibitor bortezomib and the immunomodulatory drugs (IMiDs) thalidomide and lenalidomide target MM cells in the bone marrow microenvironment and have improved outcomes.
EPIDEMIOLOGY AND ETIOLOGY
In the United States, it is estimated that 22,350 cases of MM will be diagnosed in 2013, with 10,710 deaths. It is a disease that affects older adults with a median age at diagnosis of 66 years. MM occurs more frequently in males and African Americans.3 Additionally, individuals with a first-degree relative with MM have a 3.7-fold increased risk of developing this malignancy than those with unaffected relatives.4,5
Epidemiologic data from the United States have demonstrated associations with MM and individuals who work in agriculture.6 Studies have shown an increased incidence of lymphohematopoietic cancers associated with lifetime exposure to alchalor, a commonly used pesticide. Other occupational groups associated with the development of MM include miners, carpenters and wood workers, sheet-metal workers, and furniture makers.1,4 Radiation exposure has also been historically linked to the development of MM, but existing evidence is inconclusive.7
Although the pathogenesis of MM has not been fully elucidated and the role of antigen stimulation in the pathogenesis of the disease remains controversial, the understanding of the cellular events underlying the development of MM is becoming clearer. Decades of research and improved scientific techniques have enabled closer examination of the changes that occur during the development of normal and abnormal B cells.
PATHOPHYSIOLOGY
Multiple myeloma is a genetically heterogeneous disease that belongs to a group of related diseases called paraproteinemias that are characterized by abnormal clonal plasma cell infiltration in the bone marrow. A precursor condition called monoclonal gammopathy of undetermined significance (MGUS) is associated with monoclonal immunoglobulin in the blood (≤3 g/dL [≤30 g/L]) without clinical manifestations of the complications of MM.8,9 The conversion rate of MGUS to MM is about 1% per year. The molecular changes associated with the conversion of MGUS to MM are not clear, but genome-wide studies have identified several candidate genes associated with disease progression.2,9,10 Distinct from MGUS, which is a premalignant syndrome, smoldering MM is an asymptomatic disease with a low tumor burden and an indolent course.1,4 In patients with smoldering MM, the risk of progression is about 10% per year for the first 5 years after diagnosis, about 3% per year for the next 5 years, and about 1% per year for the next 10 years.11
Although both MGUS and smoldering MM lack the clinical features of MM, they share many of the same genetic features. A characteristic feature of MM cells is the requirement for an intimate relationship with the bone marrow microenvironment, where plasma cells are nurtured in specialized niches that maintain and promote their long-term survival.12 In early MM, the balance between apoptotic and antiapoptotic genes is disrupted with overexpression of antiapoptotic genes. As the disease progresses, a greater number of gene products that confer resistance, such as mutated p53, are overexpressed.13 Molecules such as interleukin-6 (IL-6) and the transcriptional regulator nuclear factor kappa B (NF-κ B) also stimulate clonal growth and promote resistance to therapy. Given their imprecise but important role in initiation and progression of MM, IL-6 and NF-κ B are targets for both old and new therapies.13,14
![]() MM is characterized by the accumulation of malignant plasma cells in the bone marrow and the production of a monoclonal immunoglobulin (M protein). These proteins, secreted by the malignant clone, are frequently referred to as paraproteins.1,4 Both MM and normal plasma cells are produced from differentiated B cells after antigen stimulation. Whereas normal plasma cells will die within days to weeks after differentiation, MM plasma cells are immortalized.1,4 MM cells are seldom seen in large quantities in the peripheral blood because of their interaction with bone marrow stromal cells. This interaction between MM cells and bone marrow stroma is mediated by adhesion molecules within an abnormal bone marrow microenvironment and is required for growth and disease progression.14 Figure 113-1 shows several of the factors involved in disease pathogenesis and progression and potential targets for thalidomide, lenalidomide, pomalidomide, bortezomib, and carfilzomib.
MM is characterized by the accumulation of malignant plasma cells in the bone marrow and the production of a monoclonal immunoglobulin (M protein). These proteins, secreted by the malignant clone, are frequently referred to as paraproteins.1,4 Both MM and normal plasma cells are produced from differentiated B cells after antigen stimulation. Whereas normal plasma cells will die within days to weeks after differentiation, MM plasma cells are immortalized.1,4 MM cells are seldom seen in large quantities in the peripheral blood because of their interaction with bone marrow stromal cells. This interaction between MM cells and bone marrow stroma is mediated by adhesion molecules within an abnormal bone marrow microenvironment and is required for growth and disease progression.14 Figure 113-1 shows several of the factors involved in disease pathogenesis and progression and potential targets for thalidomide, lenalidomide, pomalidomide, bortezomib, and carfilzomib.
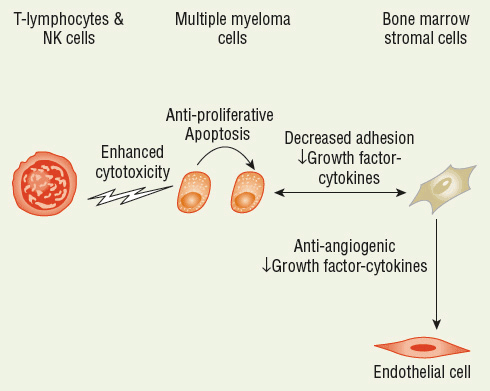
FIGURE 113-1 Sites of action for thalidomide, lenalidomide, pomalidomide, bortezomib, and carfilzomib.
Over the next several years, our current understanding of the pathogenesis of MM and the tumor-specific mutations that drive tumor development and proliferation should improve dramatically. Whole-genome sequencing may lead to improvements in clinical practice. The sequencing of the MM genome in 38 patients was recently published and revealed that mechanisms previously suspected to have a role in the biology of MM like NF-κ B may have much broader roles than previously suspected.15 Additionally, the discovery of potential new mechanisms of transformation and progression such as mutations in the oncogenic kinase BRAF may lead to new therapeutic approaches in the future.16,17
CLINICAL PRESENTATION
![]() Most patients with MM present with complaints of bone pain and fatigue at diagnosis. About 10% to 20% of patients are asymptomatic at the time of diagnosis and have what is called smoldering MM.4,11 Unfortunately, most patients show evidence of end-organ damage at the time of diagnosis. Initial laboratory evaluation often reveals hypercalcemia, renal insufficiency, anemia, and abnormalities in various disease markers, such as albumin and β2-microglobulin. Skeletal evaluation shows gross abnormalities in most patients. Bone scans show abnormalities that often include lytic lesions, osteoporosis, and fractures. This group of findings (hypercalcemia, renal insufficiency, anemia, and bone lesions) is often referred by the acronym CRAB.4,8 A bone marrow biopsy with 10% or more plasma cells and an M-protein spike on plasma or urine electrophoresis confirms the diagnosis.8,18 Immunofixation is more sensitive and identifies the M-protein isotype being secreted. In a minority of patients, no M protein can be detected in the plasma but is found in the urine, requiring that urine be examined as part of a complete diagnostic workup. About 60% of patients have intact monoclonal immunoglobulin G (IgG), 20% have monoclonal IgA, and the remaining 20% secrete only monoclonal light chains. Antibodies are composed of two light chains where antigen binds and two heavy chains. Light-chain immunoglobulin alone can be secreted by the MM clone. Free monoclonal light chains in the urine are called Bence Jones proteins because they were first described by Dr. Henry Bence Jones and are primarily responsible for MM-associated renal failure.1,4 In addition, serum IgG light chain can be measured with a free light chain assay (Freelite). This assay has several advantages compared with serum protein and urine electrophoresis, particularly increased sensitivity, and the free light chain ratio that may add valuable information on likelihood of disease progression.19
Most patients with MM present with complaints of bone pain and fatigue at diagnosis. About 10% to 20% of patients are asymptomatic at the time of diagnosis and have what is called smoldering MM.4,11 Unfortunately, most patients show evidence of end-organ damage at the time of diagnosis. Initial laboratory evaluation often reveals hypercalcemia, renal insufficiency, anemia, and abnormalities in various disease markers, such as albumin and β2-microglobulin. Skeletal evaluation shows gross abnormalities in most patients. Bone scans show abnormalities that often include lytic lesions, osteoporosis, and fractures. This group of findings (hypercalcemia, renal insufficiency, anemia, and bone lesions) is often referred by the acronym CRAB.4,8 A bone marrow biopsy with 10% or more plasma cells and an M-protein spike on plasma or urine electrophoresis confirms the diagnosis.8,18 Immunofixation is more sensitive and identifies the M-protein isotype being secreted. In a minority of patients, no M protein can be detected in the plasma but is found in the urine, requiring that urine be examined as part of a complete diagnostic workup. About 60% of patients have intact monoclonal immunoglobulin G (IgG), 20% have monoclonal IgA, and the remaining 20% secrete only monoclonal light chains. Antibodies are composed of two light chains where antigen binds and two heavy chains. Light-chain immunoglobulin alone can be secreted by the MM clone. Free monoclonal light chains in the urine are called Bence Jones proteins because they were first described by Dr. Henry Bence Jones and are primarily responsible for MM-associated renal failure.1,4 In addition, serum IgG light chain can be measured with a free light chain assay (Freelite). This assay has several advantages compared with serum protein and urine electrophoresis, particularly increased sensitivity, and the free light chain ratio that may add valuable information on likelihood of disease progression.19
As discussed earlier, the skeleton is involved at the time of diagnosis in most patients with MM.4,8 The effects of MM on the skeleton result from the abnormal production of cytokines, including IL-1, IL-6, tumor necrosis factor-α (TNF-α), and the receptor for activation of NF-κ B ligand (RANK-L). Bone disease is the net effect of the activation of osteoclasts and inhibition of osteoblastogenesis.20 In addition, patients are frequently anemic from infiltration of the bone marrow with the MM clone and poor erythropoietin response. Patients can have clinically important hypercalcemia, which results from calcium mobilization from the bone. Renal failure can occur as a result of high protein load from the monoclonal protein secretion as well as dehydration.
CLINICAL PRESENTATION Multiple Myeloma
STAGING AND PROGNOSTIC FACTORS
Some patients with MM are asymptomatic and have no evidence of end-organ damage at the time of diagnosis. As discussed previously, these patients are categorized as having smoldering (asymptomatic) MM.21 Most patients have evidence of end-organ damage (hypercalcemia [>10.5 g/dL (>2.63 mmol/L)], renal impairment [>2.0 mg/dL (>177 μmol/L)], anemia [<10 g/dL (<100 g/L; 6.21 mmol/L) or >2 g/dL (>20 g/L; 1.24 mmol/L) below normal]), or bone disease at the time of diagnosis and are categorized as having active (symptomatic) disease. Patients with asymptomatic disease have an indolent course with a median survival time of about 5 years.11,21
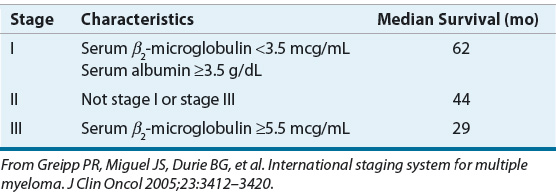
The International Staging System (ISS) uses serum β2-microglobulin and albumin concentrations to stage patients.22 These two routine laboratory tests are powerful prognostic discriminators. The ISS predicts survival in patients treated with either conventional treatment or autologous hematopoietic stem cell transplantation (HSCT). An older staging system, Durie-Salmon, uses hemoglobin, serum calcium, bone involvement, and M protein to categorize patients in one of three stages. Table 113-1 describes the ISS and median survival times for each of the three ISS stages.
TABLE 113-1 The International Staging System for Multiple Myeloma
Several adverse prognostic factors have been proposed for MM, including chromosome 13 deletion and other cytogenetic abnormalities (e.g., 17p deletion, t(4,14)), elevated β2-microglobulin, elevated C-reactive protein, high plasma cell labeling index, low albumin, and high bone marrow microvessel density.1,4,8 These prognostic factors generally represent the underlying pathologic changes associated with MM, including genetic damage (chromosome 13 and 17 abnormalities), proinflammatory changes (C-reactive protein), tumor load (β2-microglobulin), and dysregulated cellular growth (labeling index and marrow microvessel density).
TREATMENT
Desired Outcomes
![]() The current goal of therapy in MM is to prolong progression-free survival (PFS) and overall survival and improve quality of life. The initial goal of induction therapy in newly diagnosed patients with more active (symptomatic) and advanced disease (stages II and III) is to obtain at least a major response.4,8,18 This is usually followed by consolidation and maintenance therapy, both of which can extend and often improve induction responses. With the integration of novel agents into therapy, PFS and overall survival have steadily improved, and responses have increased in frequency, depth, and duration. Unfortunately, there is no convincing evidence that patients are cured of their disease.
The current goal of therapy in MM is to prolong progression-free survival (PFS) and overall survival and improve quality of life. The initial goal of induction therapy in newly diagnosed patients with more active (symptomatic) and advanced disease (stages II and III) is to obtain at least a major response.4,8,18 This is usually followed by consolidation and maintenance therapy, both of which can extend and often improve induction responses. With the integration of novel agents into therapy, PFS and overall survival have steadily improved, and responses have increased in frequency, depth, and duration. Unfortunately, there is no convincing evidence that patients are cured of their disease.
General Approach
In asymptomatic patients with smoldering MM, watchful waiting is the most common practice despite a systematic review that suggests early treatment with chemotherapy slows disease progression and may decrease vertebral compression.11,21 The benefits of chemotherapy in this setting are generally offset by the absence of convincing evidence that early treatment improves overall survival and the risk of treatment-related adverse events. With the availability of new novel agents, progression of this form of MM may be delayed. The National Comprehensive Cancer Network (NCCN) guidelines currently recommend watchful waiting for smoldering MM.23
Initial management of symptomatic MM depends on the presence or absence of high-risk features of the disease (i.e., cytogenetics), patient age, renal function, performance status, and whether autologous HSCT is planned. Although current treatments are not curative, the median survival time has increased significantly from about 7 months to 24 to 36 months in high-risk disease patients and 6 to 7 years or more in patients with standard-risk disease, primarily as a result of improved treatment of symptomatic MM and supportive care.4,23
All patients with symptomatic MM are treated with initial induction therapy. Although there is no standard initial or induction therapy, the regimens differ depending on whether the patient is a candidate for autologous HSCT (Table 113-2). The age restriction for autologous HSCT has changed because of low transplant-related mortality, but autologous HSCT is generally reserved for patients younger than 65 years of age.
TABLE 113-2 Drug Therapy in Newly Diagnosed Multiple Myeloma
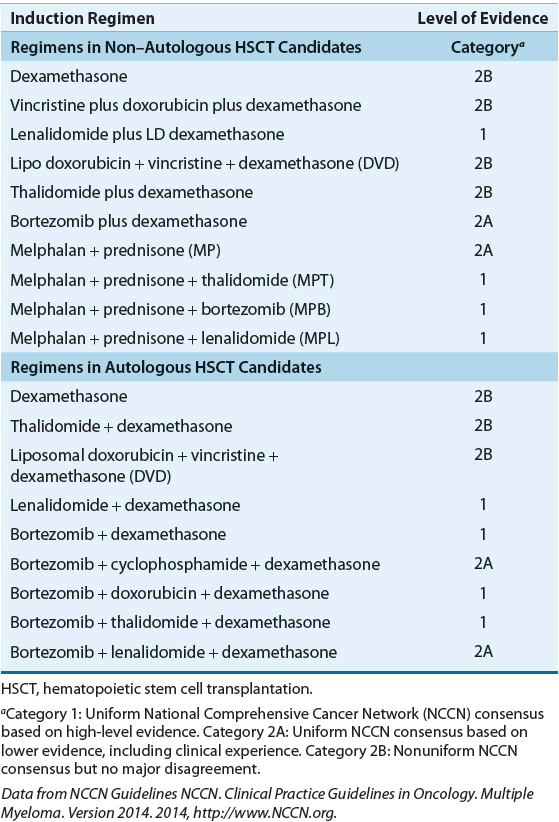
For many years, the choice of induction therapy in autologous HSCT candidates included VAD (vincristine, doxorubicin, and dexamethasone) as the standard therapy. In the last 10 years, combination regimens such as dexamethasone combined with thalidomide, lenalidomide, or bortezomib and dexamethasone combined with bortezomib and lenalidomide or thalidomide have become common. The use of VAD chemotherapy before autologous HSCT is now obsolete given data that suggest superior outcomes in patients receiving newer drug combinations.23,24
The Mayo Clinic recommends a risk-adapted approach to initial therapy in which treatment is guided by cytogenetics and gene expression profiling.25 In contrast to the single institution guidelines of Mayo Clinic, the NCCN recommendations are based on the opinions of experts from many nationally recognized cancer centers (Table 113-2).23 The Mayo Clinic and NCCN guidelines both recommend the use of newer novel agents as initial therapy; these guidelines are discussed later in Recommendations for Initial Therapy section.
Induction therapy is usually continued until maximum response is achieved. Patients who are candidates for autologous HSCT then undergo hematopoietic stem cell collection. Most patients undergo autologous HSCT at that time, but some patients may decide to delay the procedure. Patients who are not candidates for autologous HSCT usually receive several cycles of consolidation therapy, although the optimal duration of therapy after maximum response is achieved is unknown. Single-agent maintenance therapy may be given in both transplant-eligible and -ineligible patients.
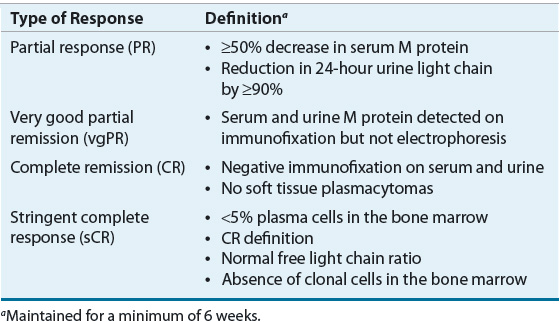
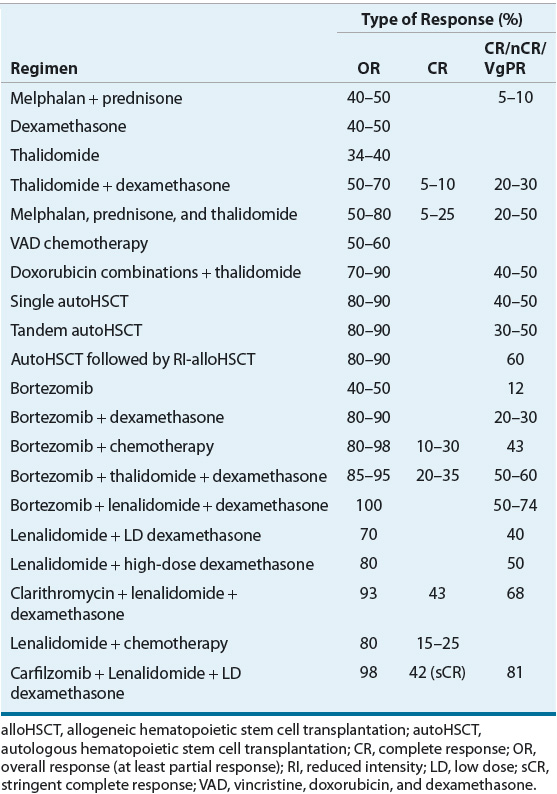
Clinical response to therapy is generally defined by a reduction in paraprotein in blood and urine.4 Clinical complete remission (CR) is defined as elimination of plasma paraprotein as measured by electrophoresis and immunofixation and plasma cells (≤5%) in the bone marrow. A specialized type of complete remission, called stringent complete response (sCR), is defined by normal free light chain and negative immunofixation. Complete remissions are uncommon in MM, and lesser responses, including partial response (PR), near complete response (nCR), and very good partial response (VGPR), are more commonly attained. Although the nCR term is less commonly used in current trials, it was used in several important studies. These lesser responses can be important because they may correlate with improved survival. Table 113-3 describes the most common types of responses that are used clinically.23
TABLE 113-3 Definition of Clinical Response in Multiple Myeloma
Pharmacotherapy of Multiple Myeloma
The current treatment of MM relies heavily on integration of novel agents, including thalidomide, lenalidomide, bortezomib, carfilzomib, and pomalidomide. These novel agents have revolutionized the treatment of MM, greatly increasing responses and survival with acceptable but different toxicity profiles compared with conventional chemotherapy-based regimens previously used in MM. Tables 113-4 and 113-5 show dosing and monitoring parameters for the novel agents used in the treatment of MM. Dose reductions in elderly patients and in patients with adverse events are often required.24
TABLE 113-4 Dosing of Novel Agents in Multiple Myeloma
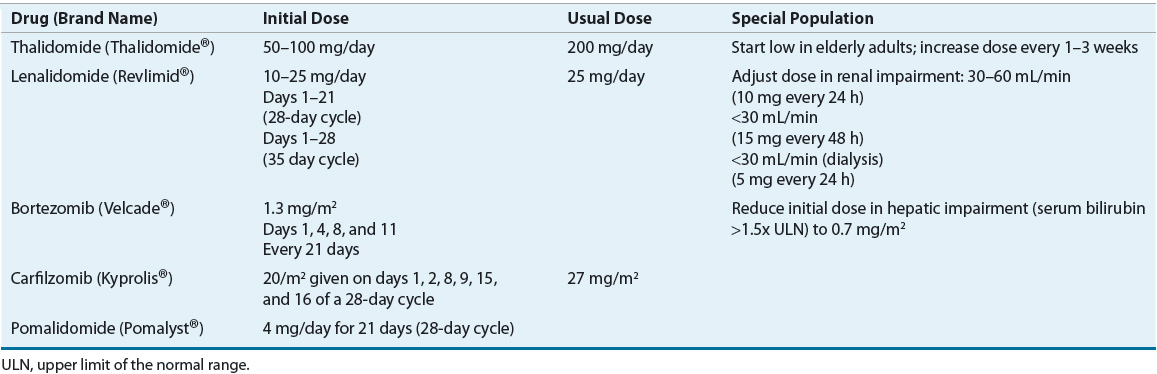
TABLE 113-5 Adverse Reactions and Monitoring Parameters for Novel Agents in Multiple Myeloma

Conventional Chemotherapy
As previously discussed, two of the common conventional chemotherapy regimens used historically to treat MM are melphalan plus prednisone (MP) and VAD.4,26 Despite more active combinations, MP and VAD remain listed as options as initial therapy in patients with MM.23 Because conventional-dose melphalan has an adverse effect on stem cell mobilization and subsequent autologous HSCT, the use of melphalan is limited to patients ineligible for autologous HSCT. Melphalan has also been associated with the development of myelodysplastic syndromes.27 The original use of VAD chemotherapy as initial treatment became more common because of these concerns with melphalan. However, the slightly higher response rates with VAD and similar combination chemotherapy did not translate into improved survival compared with MP, and VAD is now rarely used in MM.23
Because dexamethasone accounts for most of the antimyeloma activity of VAD (Table 113-6), dexamethasone was used alone as initial therapy. However, one study reported that MP produced similar response rates and survival compared with dexamethasone. The higher rate of infection and central nervous system toxicity in patients treated with dexamethasone led these investigators to conclude that high-dose dexamethasone be used with caution as initial therapy, particularly in older patients.28 In current regimens, newer agents (thalidomide, bortezomib, lenalidomide, carfilzomib) are combined with dexamethasone or the MP backbone to maximize initial response rates.4,8,23,26,29 Doxorubicin, which also is included in VAD chemotherapy, is recognized as highly active antimyeloma chemotherapy. Current regimens can combine doxorubicin in the liposomal form with various novel agents producing regimens with some of the highest responses seen in MM patients.
TABLE 113-6 Initial Therapies for Multiple Myeloma
Thalidomide (Thalomid)
Thalidomide was first used clinically in Europe in the late 1950s as a sedative and antiemetic but its use was largely abandoned when teratogenicity was reported. Its immunomodulatory effects became evident with its use in Hansen disease (or leprosy), and it continues to be used for this rare indication. These clinical benefits are thought to be related to the anti-TNF activity of thalidomide. As a result of the role of inflammatory cytokines in the pathophysiology of MM, thalidomide was first studied in refractory MM in 1999. The observation that thalidomide had activity against myeloma rejuvenated it as an important therapeutic agent.30
Thalidomide and other IMiDs have multiple immune effects, including inhibition of inflammatory mediators, antiangiogenic activity, and T cell–modulating activity. Thalidomide destabilizes TNF-α messenger RNA, which leads to increased destruction of the transcripts and reduction in TNF-α production. One potential explanation for thalidomide’s antimyeloma activity is inhibition of TNF-mediated NF-κ B activation, which results in increased apoptosis of the MM clone. Thalidomide also has TNF-independent effects on NFκ B; it protects the cytosolic inhibitor of NFκ B (Iκ B) and prevents signal transduction to the nucleus, resulting in a decline in MM growth factors.30,31
Myeloma bone marrow has a high rate of neovascularization, which makes it susceptible to antiangiogenic therapy. Bone marrow microvessel density has been identified as an independent prognostic factor in MM.32 One explanation for the angiogenesis that occurs in MM is the paracrine release of TNF-α by the myeloma clone and bone marrow stromal cells, which leads to the release of angiogenic factors, including vascular endothelial growth factor (VEGF), IL-8, basic fibroblast growth factor, and IL-1, through NF-κ B induction. Thalidomide treatment can reduce bone marrow microvessel density, which may contribute to its antimyeloma activity.
The role of TNF-α inhibition is supported by the observation that TNF-α polymorphisms may predict for thalidomide response in patients with MM.33 High producers of TNF-α had significantly higher response rates and improved survival with thalidomide therapy compared with patients without the hypersecretory phenotype. These results may be explained by inhibition of TNF-α as a required growth factor in patients with the TNF-α hypersecretory phenotype. The authors commented that larger studies are required to confirm and explain these results. Figure 113-1 shows that thalidomide inhibits proliferation and angiogenesis, stimulates T lymphocytes, and modifies the cytokine-secreting ability of bone marrow stromal cells.
Single-agent thalidomide has been extensively evaluated in refractory MM in which it produces overall response rates (including minor responses) in about 30% of patients.34 Although minor and partial responses are the most common types of responses, these end points are associated with improved survival.35
With the activity of thalidomide in refractory MM established, subsequent studies evaluated its activity in newly diagnosed patients and in combination with other therapies, including dexamethasone and chemotherapy. Partial response rates with single-agent thalidomide in untreated patients are about 30% to 40%.36 When dexamethasone is added to thalidomide in untreated patients, response rates (≥PR) increase to about 70% to 80%.37 The higher response rate with thalidomide plus dexamethasone has made this an attractive combination for initial therapy. However, the higher rate of thromboembolism with this combination (15%–20%) when used in newly diagnosed patients is a concern.37,38
![]() The addition of thalidomide to chemotherapy also increases response rates (Table 113-6). Three published randomized controlled trials in newly diagnosed MM showed that the addition of thalidomide to MP improved response.39,40 In the first randomized trial, the overall CR rate with melphalan, prednisone, and thalidomide (MPT) was about 15% compared with 4% with MP. With a median follow-up time of about 3 years, patients in the MPT group had significantly improved PFS but not overall survival.39 The second randomized trial was stopped early because MPT showed clear improvements over the other treatment arm. Results of this trial reported significantly improved median PFS (27.5 vs. 17.8 months) and overall survival (51.6 vs. 33.2 months) with MPT compared with MP.40 A third trial compared MPT with MP in patients older than 75 years of age. Patients on MPT had superior PFS and overall survival at the cost of increased peripheral neuropathy and neutropenia.41 Based on these impressive results, some have previously recommended that MPT be the new standard induction therapy in older patients ineligible for autologous HSCT. However, it is not possible to define a single standard regimen in this setting because of the number of highly active combination regimens and the lack of head-to-head comparative trials. The increased response rate of MPT is at the expense of higher rates of grades 3 and 4 toxicity, particularly venous thromboembolism (VTE), peripheral neuropathy, and infection.40,41
The addition of thalidomide to chemotherapy also increases response rates (Table 113-6). Three published randomized controlled trials in newly diagnosed MM showed that the addition of thalidomide to MP improved response.39,40 In the first randomized trial, the overall CR rate with melphalan, prednisone, and thalidomide (MPT) was about 15% compared with 4% with MP. With a median follow-up time of about 3 years, patients in the MPT group had significantly improved PFS but not overall survival.39 The second randomized trial was stopped early because MPT showed clear improvements over the other treatment arm. Results of this trial reported significantly improved median PFS (27.5 vs. 17.8 months) and overall survival (51.6 vs. 33.2 months) with MPT compared with MP.40 A third trial compared MPT with MP in patients older than 75 years of age. Patients on MPT had superior PFS and overall survival at the cost of increased peripheral neuropathy and neutropenia.41 Based on these impressive results, some have previously recommended that MPT be the new standard induction therapy in older patients ineligible for autologous HSCT. However, it is not possible to define a single standard regimen in this setting because of the number of highly active combination regimens and the lack of head-to-head comparative trials. The increased response rate of MPT is at the expense of higher rates of grades 3 and 4 toxicity, particularly venous thromboembolism (VTE), peripheral neuropathy, and infection.40,41
The combination of thalidomide, dexamethasone, and pegylated liposomal doxorubicin produces a high overall response rate of 98% and a complete remission rate of 34%. The major grades 3 and 4 toxicities were VTE (14%) and infection (22%). However, toxicity was acceptable, even in patients older than 65 years of age.42 Although the activity of doxorubicin and thalidomide compares favorably with other combinations, one disadvantage of this regimen is that pegylated liposomal doxorubicin requires IV administration.
Thalidomide dose correlates with response and toxicity. In one large trial of single-agent thalidomide, a higher response rate was observed when more than 42 g of thalidomide was administered over a 3-month period, which is equivalent to a daily dose of about 450 mg.35 As expected, the higher dose was associated with higher rates of thalidomide-related toxicity. When thalidomide is combined with chemotherapy, thalidomide doses of 100 mg/day are associated with high CR rates.39,42,43 Neuropathy, one of the important dose-limiting toxicities, may correlate with cumulative thalidomide doses. Thalidomide-induced neuropathy is usually, but not always, reversible and is associated with demyelinating changes in peripheral neurons. About 10% to 20% of patients are unable to tolerate thalidomide, and neuropathy is often the toxicity associated with discontinuation of therapy.35,41 Unfortunately, no effective methods have been identified to prevent or treat thalidomide-induced neuropathy.
Other common toxicities associated with thalidomide include constipation, sedation, and rash. Although these toxicities can be problematic, they rarely require discontinuation of thalidomide treatment. Stimulant laxatives can be used to prevent severe constipation. The severity of constipation and sedation declines over time in many patients.44
The rate of VTE with single-agent thalidomide is relatively low (<5%) and may not exceed the baseline incidence for MM patients. VTE prophylaxis is not recommended in patients receiving single-agent thalidomide.44 When thalidomide is combined with dexamethasone, MP, or doxorubicin, the risk of thrombosis is elevated. The underlying mechanism for thrombosis in these patients is unknown, but rates in several studies of combination therapy were as high as 10% to 30%.39–41,45 VTE prophylaxis is recommended and potential preventive strategies include therapeutic doses of warfarin, fixed-dose warfarin, low-molecular-weight heparin (LMWH), or aspirin depending on the patient’s risk for VTE. Fixed-dose warfarin and 100 mg aspirin was recently compared with LMWH in MM patients on thalidomide combinations. Fixed-dose warfarin and 100 mg aspirin showed similar efficacy to LMWH in lowering a composite measure of VTE and cardiac events. However, when only grade 3 to 4 VTEs were evaluated, aspirin prophylaxis was similar to LMWH, but fixed-dose warfarin was inferior.46 Warfarin is not a popular choice for VTE prophylaxis because fixed-dose warfarin remains controversial, and therapeutic warfarin is associated with bleeding complications. The evidence suggests low-dose aspirin is effective prophylaxis, but it should be reserved for patients in whom LMWH is not feasible and in whom there is a low to moderate risk of developing VTE.45–47
Bortezomib (Velcade®)
![]() Bortezomib is a proteasome inhibitor approved for use in newly diagnosed and relapsed or refractory MM. The proteasome is a protease complex responsible for degrading cytosolic proteins that are conjugated to ubiquitin. Ubiquitin is a 8.5-kD polypeptide that tags various proteins for destruction.48 By reversibly binding to the chymotrypsin site in the catalytic core of the 26S proteasome, bortezomib inhibits the degradation of these targeted proteins.
Bortezomib is a proteasome inhibitor approved for use in newly diagnosed and relapsed or refractory MM. The proteasome is a protease complex responsible for degrading cytosolic proteins that are conjugated to ubiquitin. Ubiquitin is a 8.5-kD polypeptide that tags various proteins for destruction.48 By reversibly binding to the chymotrypsin site in the catalytic core of the 26S proteasome, bortezomib inhibits the degradation of these targeted proteins.
In MM, NF-κ B activity is increased, resulting in increased transcription of inflammatory cytokines such as IL-6 and TNF-α, which are involved in the pathogenesis and progression of MM. In the cytosol, NF-κ B is bound to and inhibited by Iκ B. The proteasome degrades Iκ B. When the proteasome is inhibited with bortezomib, cytosolic concentrations of Iκ B remain high, and NF-κ B is retained in the cytosol as an inactive complex. The resulting inhibition of the NF-κ B signal leads to a reduction in cytokine production and growth inhibition of the MM clone. Other proteins involved in cell-cycle regulation and apoptotic signaling that may be affected by bortezomib include p53, JNK proteins, and caspase 3.48
In phase I studies in patients with refractory hematologic malignancies, bortezomib was administered twice weekly for 2 consecutive weeks followed by 1 week of rest. The responses observed in those studies included a CR in one of eight patients who completed the first course of therapy and minor responses in two patients. These responses were impressive for a phase I trial and confirmed the promising activity in preclinical studies.49
Patients with refractory MM were then enrolled in a phase II trial and received 1.3 mg/m2 of bortezomib twice weekly for 2 weeks followed by 1 week of rest. Patients received up to 8 cycles. The overall response rate was 35% (includes minor responses) with seven (3.6%) patients achieving a CR.50 Based on the phase I and II studies, bortezomib was approved in May 2003 under the Food and Drug Administration’s (FDA’s) accelerated approval process for relapsed or refractory MM in patients who had failed at least two prior therapies.
Subsequently, a large phase III study (Assessment of Proteasome Inhibition for Extending Remissions [APEX] trial) demonstrated that bortezomib had superior activity compared with high-dose dexamethasone in relapsed MM.51 Bortezomib-treated patients had higher complete and partial response rates (38% vs. 18%), longer median time to progression (6.2 vs. 3.5 months), and improved 1-year overall survival (80% vs. 66%) compared with patients receiving dexamethasone. The differences in each of these end points were statistically significant. The results from this study led to expanded FDA approval in 2005 to include patients who had relapsed after one therapy.
Combination therapy with bortezomib has shown promising results in relapsed MM. It was reported that relapsed patients who had suboptimal response to bortezomib alone may respond after the addition of dexamethasone. Subsequent studies reported improved results with the combination of bortezomib and corticosteroids with the CR and nCR rate ranging between 5% and 15%.52 The inclusion of bortezomib in three- to four-drug combinations, which may include doxorubicin, melphalan, thalidomide, and lenalidomide, produce CR and nCR rates of 10% to 50% in relapsed MM.48,52
A number of studies have investigated bortezomib in newly diagnosed patients (Table 113-6). Bortezomib alone produces about a 40% response rate with about 3% of patients obtaining a complete remission. When combined with dexamethasone, the overall response rate increases to about 90% (CR plus PR) with CR rates of 5% to 20%.53,54 In a phase II study of bortezomib combined with MP (MPB) in newly diagnosed elderly MM patients, the overall response rates of 89% and CR rates of 32% are among the highest reported rates with induction therapies.55 Subsequently, MPB was compared with MP in the large phase III VISTA (Velcade as Initial Standard Therapy in multiple myeloma) trial. The overall response and CR rates, time to progression, and overall survival were significantly better in the MPB group. Based on these results, bortezomib received FDA approval in 2008 as first-line therapy in newly diagnosed patients with MM. The improvement in response came at the expense of greater serious adverse effects, including neuropathy, gastrointestinal toxicity, and herpes zoster. However, treatment-related mortality was not different between MPB and MP groups.56 An update of this study reported a continued survival benefit after 5 years of follow-up.57
Bortezomib can cause significant toxicity, the most common being mild to moderate fatigue and gastrointestinal toxicities. Neuropathy occurs frequently and is the most common cause of discontinuation of therapy. In the VISTA trial, the rate of neuropathy was 44% in the MPB group versus 5% in the MP group.56 However, the MPB and MP groups had similar rates of therapy discontinuation at about 15%. Other important toxicities included thrombocytopenia, fever, neutropenia, and infection. An increased risk of shingles has been reported in bortezomib-treated patients, and the NCCN guidelines recommend that herpes zoster prophylaxis be considered.23 VTE prophylaxis is not required with bortezomib when it is combined with MP based on the results of the VISTA trial, which reported low rates of VTE and nearly identical rates in the MPB versus MP group.56
Bortezomib plus dexamethasone has more recently been added to cyclophosphamide or lenalidomide, resulting in high response rates and improved PFS.25 The use of bortezomib has become more convenient with the new subcutaneous regimens.58 In a phase III trial in relapsed MM, therapeutic equivalence was found between IV and subcutaneous routes of administration.58,59 In addition, subcutaneous administration offers the potential advantage of administration in patients without IV access and perhaps improved safety profile, particularly less peripheral neuropathy.
Lenalidomide (Revlimid®)
![]() Lenalidomide is a thalidomide analog that shares a similar mechanism of action with thalidomide but is significantly more potent. Because of differences in the toxicity profile compared with thalidomide, the use of lenalidomide has increased. In phase I studies, patients with relapsed, refractory MM were found to have a maximum tolerated dose of lenalidomide of 25 mg/day, and this dose was the most commonly used dose in subsequent phase II and III studies.60
Lenalidomide is a thalidomide analog that shares a similar mechanism of action with thalidomide but is significantly more potent. Because of differences in the toxicity profile compared with thalidomide, the use of lenalidomide has increased. In phase I studies, patients with relapsed, refractory MM were found to have a maximum tolerated dose of lenalidomide of 25 mg/day, and this dose was the most commonly used dose in subsequent phase II and III studies.60
The addition of lenalidomide to high-dose dexamethasone has been shown to increase response rate and prolong survival in patients with relapsed MM. In 2006, lenalidomide received FDA approval in relapsed-refractory MM based on the results of two randomized controlled trials.61,62 One trial was conducted in North America, and the other trial was conducted outside of North America. In both trials, patients were randomized to receive a combination of either lenalidomide (25 mg/day on days 1 to 21 of a 28-day cycle) and high-dose dexamethasone or an identical lenalidomide placebo and high-dose dexamethasone. In the North American trial, patients in the lenalidomide and dexamethasone group had overall and CR rates of 61% and 14% compared with 20% and 0.6% in the dexamethasone alone group (P < 0.001).61 These improved response rates translated into longer median overall survival time in the lenalidomide and dexamethasone group (29.6 vs. 20.5 months). Similar results were reported in the trial conducted outside of the United States.62
Lenalidomide has been extensively studied as initial therapy in newly diagnosed MM (Table 113-6). Preliminary results of phase I and phase II studies of lenalidomide plus dexamethasone report an overall response rate of 90% and a CR rate of 18%.63 These rates may be higher than those reported with thalidomide plus dexamethasone. Lenalidomide causes less neurotoxicity and constipation but more myelosuppression than thalidomide.64 When used as part of combination therapy, the risk of VTE with lenalidomide is similar to that observed with thalidomide, and VTE prophylaxis is recommended. Results from a phase III trial in newly diagnosed MM reported that patients randomized to lenalidomide plus high-dose dexamethasone had a 26% incidence of VTE compared with a 12% rate in those randomized to the lenalidomide plus low-dose dexamethasone arm.65,66 That trial also reported a superior 2-year overall survival rate in the lenalidomide plus low-dose dexamethasone group (87% vs. 75%), and this regimen could become the new standard induction regimen for older patients ineligible for autologous HSCT. The improved survival in the low-dose dexamethasone arm is related to lower mortality from adverse events, particularly VTE. Excess deaths in the high-dose dexamethasone group usually occurred in the first 4 months and in elderly patients. The low risk of VTE in the lenalidomide plus low-dose dexamethasone arm may allow for VTE prophylaxis with low-dose aspirin alone.65,66 These results have led to a category 1 NCCN recommendation in MM patients not eligible for transplant.23
Carfilzomib (Kyprolis®)
![]() Carfilzomib is the first second-generation proteasome inhibitor to receive accelerated approval from the FDA in July 2012 as treatment for patients with MM who have received at least two prior therapies, including bortezomib and an immunomodulatory agent, and have demonstrated disease progression on or within 60 days of the completion of the last therapy.
Carfilzomib is the first second-generation proteasome inhibitor to receive accelerated approval from the FDA in July 2012 as treatment for patients with MM who have received at least two prior therapies, including bortezomib and an immunomodulatory agent, and have demonstrated disease progression on or within 60 days of the completion of the last therapy.
The schedules for administration of carfilzomib are based on the results of preclinical and phase I and II studies and are different from those for bortezomib. Based on studies in preclinical murine models, which showed more potent, yet tolerable, proteasome inhibition with two consecutive daily doses, carfilzomib entered phase I clinical testing with two schedules: 5 consecutive days of 14-day cycle and 2 consecutive days weekly of a 28-day cycle.67–69 The schedule that used 2 consecutive days of dosing showed better tolerability and allowed increases in carfilzomib doses. A subsequent multivariate analysis of three phase II trials showed a dose–response relationship in MM patients. The initial study used a 20 mg/m2 fixed dose in every cycle and showed fourfold less response when compared with increasing to 27 mg/m2 in the following cycle in patients who could tolerate the initial dose.70 Collectively, subsequent clinical trials have generally adopted the administration schedule of day 1, 2, 8, 9, 15, 16, 22, and 23 of 28-day cycles with carfilzomib, starting at 20 mg/m2 IV over 2 to 10 minutes on the first cycle/week and increased to 27 mg/m2 or more afterward depending on tolerability. In addition, the results from phase Ib/II studies showed that prolonged infusion (30 min) is better tolerated and that the carfilzomib dose can be increased up to 56 mg/m2.71
The most mature safety data for carfilzomib comes from the compiled results of three phase II studies. The most frequently reported adverse events included fatigue (55%), anemia (47%), nausea (45%), thrombocytopenia (36%), dyspnea (35%), diarrhea (33%), and pyrexia (30%). The most common grade 3 or greater adverse events were thrombocytopenia (23%), anemia (22%), lymphopenia (18%), pneumonia (11%), and neutropenia (10%). Grade 2 elevation of creatinine was reported in 25% of patients and was improved with the use of hydration and dexamethasone as premedication. Most of these events were manageable.72
The single-agent activity of carfilzomib is based on phase II studies of 266 relapsed and refractory MM patients who had received a median of five previous therapies.73 Patients received carfilzomib 20 mg/m2 IV over 2 to 10 minutes twice weekly on 2 consecutive days with dexamethasone premedication for 3 of 4 weeks in cycle 1 and then 27 mg/m2 in subsequent cycles until disease progression, unacceptable toxicity, or completion of a maximum of 12 cycles. The primary end point of overall response rate (≥PR) was 22.9%, and the median duration of response was 7.8 months (95% confidence interval [CI] 5.6–9.2 months). In patients who were refractory or intolerant to both bortezomib and lenalidomide, 37% obtained clinical benefit. In patients refractory to both bortezomib and lenalidomide, the overall response rate (≥PR) was 15.4%. Moreover, unfavorable cytogenetic characteristics did not appear to adversely impact response rates. The median overall survival time was 15.6 months compared with the median of 9 months typically seen in this setting. An additional large multicenter trial in relapsed/refractory MM patients investigated variable dosing in bortezomib-naïve patients and those previously treated with bortezomib; the overall response rate reported as 52.2% in bortezomib-naïve patients in the 20/27 mg/m2 dose cohort.74 Importantly, these studies demonstrated that carfilzomib had a rapid time to response (0.5–1.0 months).74,75
The activity of carfilzomib combination regimens as first-line treatment is impressive. A phase I/II study of carfilzomib in combination with lenalidomide and dexamethasone enrolled 53 newly diagnosed patients treated with carfilzomib 20, 20/27, or 20/36 mg/m2 in phase I and expansion to phase II at a 20/36 mg/m2 dose.76 The overall response rate (≥PR) was 94%. The responses were rapid and increased in depth with additional cycles of therapy, with 62% and 42% of patients achieving a CR and sCR, respectively. In 36 patients who completed induction, 78% reached at least nCR and 61% sCR. At a median follow-up time of 13 months, the estimated 24-month PFS was 92%. The three-drug regimen did not adversely affect stem cell collection, but was associated with peripheral neuropathy, which was predominately grade 1 or 2 and observed in 23% of patients.
Three randomized phase III trials are ongoing in relapsed myeloma. The CArfilzomib, Lenalidomide, and DexamethaSone versus Lenalidomide and Dexamethasone for the treatment of PatIents with Relapsed Multiple MyEloma (ASPIRE) trial compares lenalidomide and dexamethasone with lenalidomide, dexamethasone, and carfilzomib; the CarFilzOmib for AdvanCed Refractory MUltiple Myeloma European Study (FOCUS) trial compares carfilzomib monotherapy to best supportive care; and the RandomizEd, OpeNLabel, Phase 3 Study of Carfilzomib Plus DExamethAsone Vs Bortezomib Plus DexamethasOne in Patients with Relapsed Multiple Myeloma (ENDEAVOR) trial compares carfilzomib and dexamethasone with bortezomib and dexamethasone.
Drugs in Development
![]() Pomalidomide is the newest in the immunomodulatory class of antimyeloma drugs. It was recently granted accelerated approval by the FDA in relapsed MM. In a phase II trial, the combination of pomalidomide with dexamethasone produced good overall response rates (35%) in heavily pretreated relapsed and refractory MM. Toxicity profile was reasonable and consisted mainly of manageable myelosuppression. Pomalidomide is currently being evaluated in phase III trials.77
Pomalidomide is the newest in the immunomodulatory class of antimyeloma drugs. It was recently granted accelerated approval by the FDA in relapsed MM. In a phase II trial, the combination of pomalidomide with dexamethasone produced good overall response rates (35%) in heavily pretreated relapsed and refractory MM. Toxicity profile was reasonable and consisted mainly of manageable myelosuppression. Pomalidomide is currently being evaluated in phase III trials.77
Recommendations for Initial Therapy
![]() The Mayo Clinic Guidelines uses a risk-adapted approach that categorizes patients into risk groups based on cytogenetics and gene expression profiling. In high-risk patients, the combination of bortezomib, lenalidomide, and dexamethasone is recommended as induction therapy.25 In intermediate-risk patients, the combination of bortezomib, cyclophosphamide, and dexamethasone is recommended as induction therapy. In both high- and intermediate-risk patients, induction therapy for 4 months is recommended in transplant-eligible patients and for 1 year in transplant-ineligible patients. In the largest group, standard-risk patients, lenalidomide and low-dose dexamethasone or bortezomib, cyclophosphamide, and dexamethasone are recommended for 4 cycles in transplant-eligible patients followed by transplant, but transplant can be delayed depending on patient preference. Transplant-ineligible standard-risk patients should receive lenalidomide and low-dose dexamethasone, with dexamethasone dose reduction or discontinuation after 1 year Other options for induction therapy in these patients is MPT or a bortezomib-based regimen (in patients with renal insufficiency). Many patients receive maintenance therapy with bortezomib or lenalidomide after transplant (in transplant-eligible patients) or induction therapy (in transplant-ineligible patients).
The Mayo Clinic Guidelines uses a risk-adapted approach that categorizes patients into risk groups based on cytogenetics and gene expression profiling. In high-risk patients, the combination of bortezomib, lenalidomide, and dexamethasone is recommended as induction therapy.25 In intermediate-risk patients, the combination of bortezomib, cyclophosphamide, and dexamethasone is recommended as induction therapy. In both high- and intermediate-risk patients, induction therapy for 4 months is recommended in transplant-eligible patients and for 1 year in transplant-ineligible patients. In the largest group, standard-risk patients, lenalidomide and low-dose dexamethasone or bortezomib, cyclophosphamide, and dexamethasone are recommended for 4 cycles in transplant-eligible patients followed by transplant, but transplant can be delayed depending on patient preference. Transplant-ineligible standard-risk patients should receive lenalidomide and low-dose dexamethasone, with dexamethasone dose reduction or discontinuation after 1 year Other options for induction therapy in these patients is MPT or a bortezomib-based regimen (in patients with renal insufficiency). Many patients receive maintenance therapy with bortezomib or lenalidomide after transplant (in transplant-eligible patients) or induction therapy (in transplant-ineligible patients).
Initial therapy in the NCCN guidelines is based on transplant eligibility. In patients ineligible for autologous HSCT, thalidomide, lenalidomide, or bortezomib is added to chemotherapy as initial therapy. MP forms the backbone to which these newer drugs are added if the patient is not a candidate for autologous HSCT. As previously discussed, MPT produces high response rates, and MPB produce equal or better results.37 Based on the results of phase III trials demonstrating superiority of MPT over MP, many suggested that MPT was the preferred induction regimen in patients who were ineligible for autologous HSCT. However, the results of the VISTA trial, the continuous lenalidomide trial, in which MPL induction is followed by lenalidomide maintenance, and a phase II trial comparing MP with MPL in newly diagnosed patients supports the addition of MPB and MPL as two additional preferred induction regimens.56,78,79 MPB, MPL, and MPT are listed as NCCN category 1 recommendations.23 Bortezomib-containing regimens (i.e., MPB) may be particularly useful in MM patients with high-risk cytogenetics (t(4;14), 17p-). The preferred combination (MPT, MPB, or MPL) is currently unclear and will require randomized controlled trials that compare these combinations. Carfilzomib-based therapy will have an important role in heavily pretreated refractory MM and is being evaluated in ongoing phase III trials as induction therapy for newly diagnosed MM patients.
If autologous HSCT is planned after induction therapy, melphalan should be avoided, and thalidomide, bortezomib, or lenalidomide can be added to dexamethasone or VAD-like chemotherapy. The NCCN guidelines list several induction therapy options (Table 113-2). Because there is no standard induction regimen, clinicians can select from a wide range of possible induction regimens.29 Many clinicians recommend lenalidomide or bortezomib and dexamethasone as two-drug induction regimens or bortezomib, dexamethasone, and either cyclophosphamide, doxorubicin, or lenalidomide as three-drug regimens for patients who are autologous HSCT candidates.
![]() Patients with high-risk cytogenetics may benefit from bortezomib-containing induction regimens because of its activity in these high-risk patients.14,25 Because patients with high-risk cytogenetics may have poorer outcomes after autologous HSCT, bortezomib-containing regimens should be considered in this group of patients.14,25
Patients with high-risk cytogenetics may benefit from bortezomib-containing induction regimens because of its activity in these high-risk patients.14,25 Because patients with high-risk cytogenetics may have poorer outcomes after autologous HSCT, bortezomib-containing regimens should be considered in this group of patients.14,25



