The evidence for the essentiality of four ultratrace elements—cobalt, iodine, molybdenum, and selenium—is substantial and noncontroversial; specific biochemical functions have been defined for these elements. Iodine and selenium are recognized as elements of major nutritional importance and are the topics of Chapters 38 and 39. Although cobalt is required in ultratrace amounts, it has to be in the form of vitamin B12; therefore cobalt as part of vitamin B12 is covered in Chapter 25. Very little nutritional attention is given to molybdenum because a deficiency has not been unequivocally identified in humans other than in an individual nourished by total parenteral nutrition or in individuals with rare genetic defects that cause metabolic disturbances involving the functional roles of this element. Therefore molybdenum is discussed in this chapter. Molybdenum is a transition element that readily changes its oxidation state and can thus act as an electron transfer agent in oxidation–reduction reactions in which it cycles from Mo6+ to reduced states. This explains why molybdenum functions as an enzyme cofactor in all forms of life. Molybdenum is present at the active site in a small nonprotein cofactor containing a pterin nucleus in all known molybdoenzymes except nitrogenase (Johnson, 1997). More than 40% of the molybdenum not attached to an enzyme in the liver exists as this cofactor bound to the mitochondrial outer membrane. This form is transferred to an apomolybdoenzyme to make the holomolybdoenzyme (Rajagopalan, 1988). The molybdoenzymes in mammalian systems also contain Fe–S centers and flavin cofactors. Molybdoenzymes catalyze the hydroxylation of various substrates by using oxygen atoms from water. Aldehyde oxidase oxidizes and detoxifies various pyrimidines, purines, pteridines, and related compounds. Xanthine oxidase/dehydrogenase catalyzes the transformation of hypoxanthine to xanthine and of xanthine to uric acid. Hypoxanthine and xanthine are intermediates in the degradation of purines. Sulfite oxidase catalyzes the transformation of sulfite, formed mainly from cysteine catabolism, to sulfate. Essential Elements The concept of essential nutrients was well established by 1940 with a definition of food substances necessary for life, tissue growth, and repair that the body cannot synthesize. Essential nutrients were identified when dietary deficiency led to disease or failure to grow. In the 1960s and 1970s, new criteria appeared for establishing essentiality of mineral elements that could not be fed at dietary concentrations low enough to cause death or interrupt the life cycle (growth, development, or maturation such that procreation is prevented). The major criterion was that a dietary deficiency had to consistently and adversely change a biological function from optimal, and this change was preventable or reversible by physiological amounts of the element. In the 1980s and 1990s, establishing essentiality on the basis of this criterion began to receive resistance when many elements were suggested to be essential based on some small change in a physiological or biochemical variable after experimental animals were supposedly fed a diet deficient in an element. These elements included those in Table 41-1. Doubts arose about whether many of the changes were really the result of suboptimal functions. It was suggested that many of the changes were the result of pharmacological or supranutritional amounts of an element fed to supplemented controls having a beneficial effect through overcoming the lack of some other nutrient, preventing a detrimental effect caused by an inappropriate intake of another dietary substance, or affecting intestinal organisms. As a result, if the lack of an element cannot be shown to cause death or interrupt the life cycle, most nutritionists today do not consider an element essential unless it has a defined biochemical function. However, several mineral elements were often described as essential, including chromium and fluoride, before current criteria came into vogue. Therefore these are still often stated as essential in the literature when they probably should be reported as possibly essential or bioactive beneficial mineral elements. broad range of intakes (Novotny and Turnland, 2006, 2007). Men absorbed 90% to 94% of daily intakes of molybdenum ranging from 22 to 1,490 μg. Molybdenum absorption was most efficient at the highest levels of dietary molybdenum. The amount and percentage of molybdenum excreted in the urine increased as dietary molybdenum increased, which suggests that urinary excretion rather than regulated absorption is the major homeostatic mechanism for molybdenum. The absorption of molybdenum can be reduced by diets high in sulfur, because the sulfate anion is a competitive inhibitor of molybdenum absorption (Mills and Davis, 1987). Molybdate is transported loosely attached to erythrocytes in blood (Johnson, 1997). Concentrations of molybdenum in tissues, blood, and milk vary with molybdenum intake. For example, mean plasma concentration for men with an intake of 22 μg/day was 0.51 μg/L, with an intake of 121 μg/day it was 1.17 μg/L, and with an intake of 1,490 μg/day it was 6.22 μg/L (Novotny and Turnland, 2007). Highest concentrations of molybdenum are found in liver, kidney, and bone (normally >1 mg/kg dry weight) (Johnson, 1997). The concentration of molybdenum in other tissues usually is between 0.14 and 0.20 mg/kg dry weight. The molybdenum cofactor is an unstable reduced pterin with a unique four-carbon side chain, which is synthesized by a complex pathway that requires the protein products encoded by at least four different genes (MOCS1, MOCS2, MOCS3, and GEPH) (Reiss and Johnson, 2003). Both MOCS1 (molybdenum cofactor synthesis-step 1) and MOCS2 (molybdenum cofactor synthesis-step 2) are bicistronic genes, encoding two proteins in different open reading frames. The protein products are designated MOCS1A, MOCS1B, MOCS2A, and MOCS2B and are required for synthesis of molybdopterin. Molybdopterin synthase (MOCS2A and MOCS2B) must be activated by a sulfotransferase that is the product of MOCS3. GEPH encodes gephyrin, which is required during cofactor assembly for insertion of molybdenum. Gephyrin is a bifunctional protein that is essential for synaptic clustering of inhibitory neurotransmitter receptors in the central nervous system as well as the biosynthesis of molybdopterin in peripheral tissues. The steps in molybdenum cofactor formation are illustrated in Figure 41-1. Molybdenum cofactor deficiency is a rare inborn error of metabolism. Mutations that affect molybdenum cofactor synthesis result in the simultaneous loss of activity of all human molybdenum cofactor–dependent enzymes. The combined deficiency of sulfite oxidase, xanthine dehydrogenase, and aldehyde oxidase results in a severe phenotype clinically similar to that in patients with the rarer isolated sulfite oxidase deficiency. The two conditions can easily be distinguished by diminished uric acid levels and elevated xanthine concentrations in plasma and urine that result from the decreased xanthine dehydrogenase activity in the combined cofactor deficiency but not in isolated sulfite oxidase deficiency. Diagnosis of molybdenum cofactor deficiency is usually made shortly after birth because of a failure to thrive and neonatal seizures that are unresponsive to therapy. The disease is associated with a pronounced and progressive loss of white matter in the brain. Biochemical diagnostic changes include elevated urinary sulfite and S-sulfocysteine, hypouricemia, elevated plasma S-sulfonated transthyretin, and deficient molybdoenzyme activity in fibroblasts. Mild cases of the disease have been reported, probably the result of a low level residual activity of the mutant protein. However, most recognized cases have been severe; most patients die in early childhood and some survive for only a few days. There currently is no effective dietary or oral therapy for molybdenum cofactor deficiency. However, a single case was successfully treated by daily intravenous administration of cyclic pyranopterin monophosphate (Veldman et al., 2010). Most disease-producing mutations have been identified in MOCS1 and MOCS2 (Reiss and Johnson, 2003). Mutations of MOCS3 are very rare (Yamamoto et al., 2003; Ichida et al., 2001). A mutation in GEPH was identified in fibroblasts of a patient from a family with three deceased children who had all been diagnosed as molybdenum cofactor–deficient (Reiss et al., 2001). Although molybdenum is an established essential element, it is not of much practical concern in human nutrition. Reports describing signs of nutritional molybdenum deficiency in humans are rare. A convincing case of molybdenum deprivation was observed in a patient with Crohn’s disease who was receiving prolonged parenteral nutrition therapy (Abumrad et al., 1981). Signs and symptoms exhibited by this patient, which were exacerbated by methionine administration, included high methionine and low uric acid levels in blood, high oxypurines and low uric acid in urine, and very low urinary sulfate excretion. This patient had mental disturbances that progressed to coma. Intravenous supplementation with ammonium molybdate improved the clinical condition, reversed the sulfur-handling defect, and normalized uric acid production. Men fed only 22 μg molybdenum/day for 102 days maintained molybdenum balance and exhibited no biochemical signs of deficiency (Turnlund et al., 1995). In a separate study, four men adapted to consuming diets that provided 22 μg daily exhibited decreased uric acid and increased xanthine excretion in urine in response to a load dose of adenosine monophosphate; these changes suggest that xanthine oxidase activity was decreased by the low-molybdenum regimen (Turnlund et al., 1995). Molybdenum deficiency signs also are difficult to induce in animals (Mills and Davis, 1987). In rats and chicks, excessive dietary tungsten was used to restrict molybdenum absorption, and thus induce molybdenum deficiency signs, which included depressed activities of molybdoenzymes, disturbed uric acid metabolism, and increased susceptibility to sulfite toxicity. Beneficial effects have been reported for supranutritional amounts of molybdenum in the form of tetrathiomolybdate, which inhibits metal transfer functions between copper trafficking proteins (Alvarez et al., 2010). Copper-lowering therapy by tetrathiomolybdate has been found to inhibit cancer growth in five rodent models, and in advanced and metastatic cancer in dogs and humans (Brewer, 2003). In animal studies, tetrathiomolybdate inhibited pulmonary fibrosis induced by bleomycin, hepatitis induced by concanavalin A, cirrhosis induced by carbon tetrachloride (Brewer, 2003), and hyperglycemia induced by streptozotocin (Zeng et al., 2008). Supranutritional amounts of sodium molybdate were found to prevent hyperinsulinemia and hypertension induced by fructose in rats (Güner et al., 2001). Although molybdenum deficiency has not been observed in healthy people, Dietary Reference Intakes (DRIs) have been set for molybdenum (Institute of Medicine [IOM], 2001). Based on the balance studies done by Turnlund and colleagues (1995), which indicated adults could achieve molybdenum balance on an intake of 22 μg/day, the requirement was estimated to be 25 μg/day (22 μg/day plus 3 μg/day to allow for miscellaneous losses not measured in the study). Because some foods, such as soy, have lower bioavailability than the foods provided in the balance studies done by Turnlund and co-workers (1995), an average bioavailability of 75% was used to set an Estimated Average Requirement (EAR) of 34 μg/day for adults. The Recommended Dietary Allowance (RDA) was set as the EAR + 30% and is 45 μg/day for adults. DRIs for children were extrapolated from the adult value, using metabolic weight (kg0.75) as the base for extrapolation. Molybdenum has relatively low toxicity. Based largely on animal studies, the IOM (2001) set the UL for molybdenum at 2,000 μg/day (or 2.0 mg/day) for adults. For most of the population, the diet is the most important source of molybdenum. Plant foods are the major sources of molybdenum in the diet, and their molybdenum content depends upon the content of the soil in which they are grown. Good food sources of molybdenum include legumes, grain products, and nuts. Two studies of molybdenum intake in the United States yielded average intakes ranging from 76 to 240 μg/day for adults (Pennington and Jones, 1987; Tsongas et al., 1980). Therefore almost all diets should meet the RDA for molybdenum. Boron has been shown to be essential for the completion of the life cycle (i.e., deficiency causes impaired growth, development, or maturation such that procreation is prevented) for organisms in all phylogenetic kingdoms (Nielsen, 2008a). In the animal kingdom, the lack of boron was shown to adversely affect reproduction and embryo development in both the African clawed frog (Xenopus laevis) and zebra fish. Experiments with mammals have not shown that the life cycle can be interrupted by boron deprivation, nor has a biochemical function been defined for it. However, substantial evidence exists for boron being a bioactive food component that is beneficial, if not required, for health (Nielsen, 2008a). Nutritional amounts of boron fed to animals consuming a low-boron diet induce biochemical and functional changes considered beneficial for bone growth and maintenance, brain function, and inflammatory response regulation. For humans, boron intakes of 1 to 3 mg/day compared to intakes of 0.25 to 0.50 mg/day apparently have beneficial effects on bone and brain health (Nielsen, 2008a). In addition, some recent epidemiological and cell culture studies suggest that boron may have a protective effect against prostate, cervical, breast, and lung cancers (Nielsen, 2008a). Ribose is a component of adenosine. The diverse actions of boron might occur through its reaction with biomolecules containing adenosine or formed from adenosine-containing precursors. S-Adenosylmethionine and diadenosine phosphates have higher affinities for boron than any other currently recognized boron ligands in animal tissues (Hunt, 2008). Diadenosine phosphates are present in all animal cells and function as signal nucleotides associated with neuronal response. S-Adenosylmethionine, which is synthesized from adenosine triphosphate and methionine, is one of the most frequently used enzyme substrates in the body (see Chapter 25). About 95% of S-adenosylmethionine is used in methylation reactions, which influences the activity of DNA, RNA, proteins, phospholipids, hormones, and neurotransmitters. The methylation reactions result in the formation of S-adenosylhomocysteine, which can be hydrolyzed into homocysteine. High circulating homocysteine and depleted S-adenosylmethionine have been implicated in many of the disorders suggested to be affected by nutritional intakes of boron, including osteoporosis, arthritis, cancer, diabetes, and impaired brain function. The suggestion that boron bioactivity may be associated with S-adenosylmethionine is supported by the finding that the bacterial quorum-sensing signal molecule, auto-inducer AI-2, is a furanosyl borate ester synthesized from S-adenosylmethionine (Chen et al., 2002). Quorum sensing is the cell-to-cell communication between bacteria accomplished through the exchange of extracellular signaling molecules (auto-inducers). Deprivation studies with rats also indicate that boron is bioactive through affecting S-adenosylmethionine. Boron deprivation increased plasma homocysteine and decreased liver S-adenosylmethionine and S-adenosylhomocysteine (Nielsen, 2009a). Boron also strongly binds oxidized nicotinamide adenine dinucleotide (NAD+) and thus may influence reactions in which NAD+ is involved. For example, extracellular NAD+ binds to the plasma membrane receptor, CD38, an adenosine diphosphate (ADP)-ribosyl cyclase that converts NAD+ to cyclic ADP ribose (ADPR) (also see Chapter 24). ADPR is released intracellularly (Eckhert, 2006) where it binds to the ryanodine receptor and releases Ca2+ from the endoplasmic reticulum. Thus boron may be bioactive through binding NAD+ and/or ADPR and inhibiting Ca2+ release. Ca2+ is a signal ion for many processes in which boron has been shown to have an effect, including insulin release, bone formation, immune response, and brain function. Boron also may become bioactive through forming diester borate complexes with phosphoinositides, glycoproteins, and glycolipids, which contain cis-hydroxyl groups, in membranes. The finding that the borate transporter NaBC1, which apparently is essential for boron homeostasis in animal cells, conducts Na+ and OH– across cell membranes in the absence of boron (Park et al., 2004) supports the suggestion that boron affects the transduction of regulatory ions across cell membranes. It is speculated that the primary essential role of boron in plants, perhaps involving interplay between boron and calcium, is at the cell membrane level that affects signaling events (Bolaños et al., 2004).
Molybdenum and Beneficial Bioactive Trace Elements
Definition of Ultratrace Elements
Molybdenum
Biochemical Forms and Physiological Actions
Absorption, Transport, and Excretion
 NUTRITION INSIGHT
NUTRITION INSIGHT
Molybdenum Cofactor Synthesis
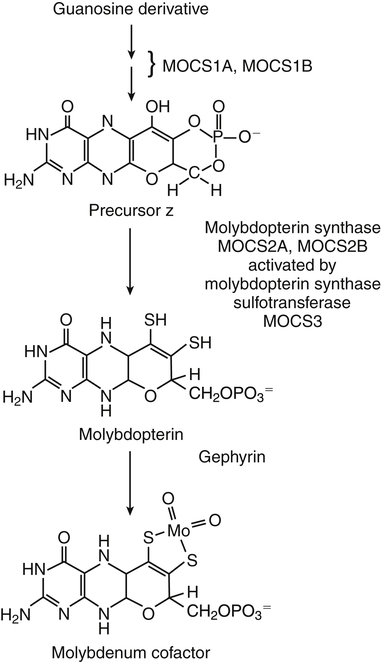
Inborn Errors in Molybdenum Cofactor Synthesis
Nutritional Requirement
Dietary Reference Intakes
Boron
Nutritional and Physiological Significance
Possible Biochemical Forms and Physiological Actions
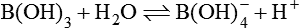
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Basicmedical Key
Fastest Basicmedical Insight Engine
 DRIs Across the Life Cycle:
DRIs Across the Life Cycle: