Tumor
Cytogenetic alteration
Fusion genes [significance]
Alveolar rhabdomyosarcoma
t(2;13)(q35;q14)
PAX3–FOXO1(FKHR)
Alveolar rhabdomyosarcoma
t(1;13)(p36;q14)
PAX7–FOXO1(FKHR) – [favorable prognosis]
Alveolar soft part sarcoma
t(X;17)(p11;q25)
TFE3–ASPSCR1(ASPL) – [sensitive to MET inhibitors]
Clear cell sarcoma
t(12;22)(q13;q12)
EWSR1–ATF1
Clear cell sarcoma
t(2;22)(q33;q12)
EWSR1–CREB1
Dermatofibrosarcoma protuberans
t(17;22)(q22;q13)
COL1A1–PDGFB
Desmoplastic small round cell tumor
t(11;22)(p13;q12)
EWSR1–WT1
Endometrial stromal sarcoma
t(7;17)(p15;q21)
JAZF1-SUZ12 [correlates with low-grade histology]
Endometrial stromal sarcoma
t(6;7)(p21;p15)
JAZF1-PHF1 [correlates with low-grade histology]
Endometrial stromal sarcoma
t(10;17)(q22;p13)
FAM22A/B-YWHAE [correlates with high-grade histology]
Epithelioid hemangioendothelioma
t(1;3)(p36;q23-24)
WWTR1–CAMTA1
Ewing/PNET
t(11;22)(q24;q12)
EWSR1–FLI1 (~80%)
Ewing/PNET
t(21;22)(q22;q12)
EWSR1–ERG
Ewing/PNET
t(7;22)(p22;q12)
EWSR1–ETV1 (rare)
Ewing/PNET
t(2;22)(q33;q12)
EWSR1–E1AF (rare)
Ewing/PNET
t(17;22)(q12;q12)
EWSR1–FEV (rare)
Infantile (congenital) fibrosarcoma
t(12;15)(p13;q25)
ETV6–NTRK3
Inflammatory myofibroblastic tumor
(2p23)
ALK rearrangements
Low-grade fibromyxoid sarcoma
t(7;16)(q34;p11)
FUS(TLS)–CREB3L2
Myxoid chondrosarcoma
t(9;22)(q22;q12)
EWSR1–NR4A3(CHN)
Myxoid chondrosarcoma
t(9;17)(q22;q11)
TAF2N–NR4A3(CHN)
Myxoid chondrosarcoma
t(9;15)(q22;q21)
TCF12–NR4A3(CHN)
Myxoid chondrosarcoma
t(3;9)(q11;q22)
TFG–NR4A3(CHN)
Myxoid/round cell liposarcoma
t(12;16)(q13;p11)
FUS(TLS)–DDIT3(CHOP)
Myxoid/round cell liposarcoma
t(12;22)(q13;q12)
EWSR1– DDIT3(CHOP)
Synovial sarcoma
t(X;18)(p11;q11)
SS18(SYT)–SSX1 – [poor prognosis]
Synovial sarcoma
t(X;18)(p11;q11)
SS18(SYT)–SSX2 – [better prognosis]
The resulting fusion gene (or transcript) is present in the majority of cases of each of these categories of sarcomas; i.e., the presence of these genetic alterations is a sensitive diagnostic test
The specific fusion gene (or transcript) for each tumor type is very often not associated with any other type of tumor; i.e., the presence of these genetic alterations is a specific diagnostic test
♦
The detection of these chromosomal translocations is often very helpful for primary diagnosis, particularly since the similar morphology and immunophenotype of many of these tumors makes definitive diagnosis by morphology and IHC alone extremely challenging
Detection of specific translocations can also yield prognostic information , as outcomes and behavior of some sarcomas can differ depending on the specific translocation or breakpoint involved in tumor formation
♦
Polymerase chain reaction (PCR)-based diagnostic methods are becoming routine – with one primer directed at each specific fusion partner
Only tumor cells with the specific translocation will yield a PCR-positive signal
Non-tumor cells (or tumor cells of a different “type”) will be PCR negative
Due to the heterogeneity of translocation breakpoints within introns, reverse transcriptase PCR (RT-PCR) using RNA (not DNA) as the typical PCR-based diagnostic target is typically employed
•
RT-PCR is also a powerful tool for minimal residual disease detection and monitoring the efficacy of therapy in low-volume specimens
♦
Fluorescence in situ hybridization (FISH) is a very useful technique for detecting chromosomal translocations, especially since the use of a break-apart probe strategy allows for detection of a rearrangement regardless of its translocation partner, unlike PCR, which requires a separate probe for each alternate partner
FISH can usually detect an abnormality when present in as few as 5% of the sampled cells
FISH is best performed on touch preparations or smears of fresh tissue, if possible: however, formalin-fixed paraffin-embedded (FFPE) tissue usually yields satisfactory results
♦
In most of these sarcomas, the fusion genes created by these specific chromosomal translocations encode transcription factors aberrantly regulating gene expression
The novel chimeric fusion proteins formed by the translocation often obtain novel biochemical functions – typically as a “new” transcription factor with altered activity
The novel biochemical activities induced by these chimeric fusion proteins include enhanced proliferative capability, inhibition of apoptosis, inhibition of differentiation, and facilitation of lineage commitment – all specific properties of tumor cells
Ewing Sarcoma/Primitive Neuroectodermal Tumor
♦
t(11;22)(q24;q12) EWSR1–FLI1
♦
t(21;22)(q22;q12) EWSR1–ERG
♦
Ewing sarcoma/primitive neuroectodermal tumor (EWS/PNET) is a tumor that typically arises in the soft tissue of children/young adults
However, the tumor has been documented in all ages and many different anatomic sites
♦
Genetically, the tumor is characterized by a balanced chromosomal translocation that results in a chimeric protein containing the EWSR1 gene product and a member of the ETS transcription factor family
Over 90% of these translocations are t(11;22)(q24;q12), but occasionally t(21;22)(q22;q12) and rarely other translocations are involved
These translocations have a variable breakpoint resulting in many structurally distinct fusion proteins
All fusion products are highly expressed and transcriptionally active, contributing to tumorigenesis
While these fusions were previously thought to be prognostic, multicenter trials failed to demonstrate any prognostic significance related to translocation
♦
Multiplex RT-PCR (with a primer directed at each specific fusion partner) has been used for the detection of chimeric genes in clinical testing with a sensitivity of 90–95%
Alternatively, FISH using break-apart probes has also been successful (Fig. 12.1)
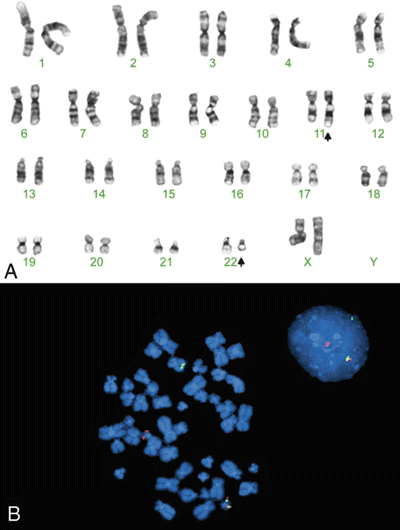
Fig. 12.1.
A reciprocal translocation [t(11;22)(q24;q12)] resulting in an EWSR1/FLI1 gene fusion in an Ewing sarcoma. (A) Shows a G-banded metaphase chromosome analysis. (B) Shows a FISH image (with a metaphase cell on the left and an interphase cell on the right). The EWSR1 “break-apart” FISH probe consists of separate green- and red-labeled probes that hybridize to a contiguous region of the EWSR1 gene on chromosome 22. In a normal cell, both probe spots are contiguous, yielding a yellow fusion signal. In an Ewing tumor cell with an EWSR1 translocation, the two probes migrate to different chromosomes (i.e., break-apart) and yield separate red and green signals. Note that this type of FISH strategy does not identify the exact partner gene involved with EWS in the translocation.
Furthermore, these highly sensitive techniques have been used in detection of minimal disease, with prognostic implications
Desmoplastic Small Round Cell Tumor
♦
t(11;22)(p13q12) EWSR1–WT1
♦
Desmoplastic small round cell tumor (DSRCT) is a sarcoma that typically occurs in young males but has been reported in both sexes of all ages
♦
Similar to EWS/PNET, DSRCT is characterized by a translocation involving the EWSR1 gene on chromosome 22
However, the fusion partner gene in this case is the Wilms’ tumor gene (WT1) on chromosome 11
Like EWS/PNET, there is variation as to the exact breakpoint
♦
By using multiplex RT-PCR, with a primer directed at each specific fusion partner, a detection sensitivity of approximately 80% has been reported in clinical samples, with a specificity near 100%
Alveolar Rhabdomyosarcoma
♦
t(2;13)(q35;q14) PAX3– FOXO1(FKHR)
♦
t(1;13)(p36;q14) PAX7– FOXO1(FKHR)
♦
Alveolar rhabdomyosarcoma (ARMS) is a round cell tumor that exhibits skeletal muscle differentiation typically occurring in young people but also less commonly in older patients
♦
Approximately 70–75% of cases have a t(2;13)(q35;q14) translocation involving the PAX3 and FOXO1(FKHR) genes
Another 10% harbor the t(1;13)(p36;q14) translocation (PAX7–FOXO1), which is associated with localized disease and a more favorable prognosis
The remaining 15–20% of so-called “fusion-negative” ARMS lesions represent a heterogeneous subgroup with variable genetic and chromosomal abnormalities, some of which involve PAX and/or FOXO1
The breakpoint always occurs in intron 1 of FOXO1 and intron 7 of PAX3/7
However, since all of these introns contain many kilobases of DNA, the exact breakpoints in a given tumor can vary over large genomic regions
The PAX3 and PAX7 gene products are involved in the regulation of normal embryonal development, and the PAX3/7–FOXO1 fusion genes appear to act as aberrant transcriptional activators
♦
Primer pairs specific for homologous regions of the PAX3/7 and FOXO1 genes have been used in PCR reactions that allow amplification of the fusion gene regardless of whether or not it involves PAX3 or PAX7
FISH has also been used successfully with excellent concordance with PCR
Synovial Sarcoma
♦
t(X;18)(p11;q11) SS18(SYT)–SSX1
♦
t(X;18)(p11;q11) SS18(SYT)–SSX2
♦
Synovial sarcoma is a spindle cell tumor predominantly occurring in deep soft tissues of the extremities in young adults
♦
Almost all cases have an t(x;18) translocation that fuses the SS18(SYT) gene with a member of the SSX gene family, SSX1 or SSX2
Tumors other than synovial sarcomas that bear this translocation have only rarely been reported
Two morphologic variants exist, a biphasic form (i.e., spindled and epithelioid components) that usually involves the SSX1 gene and a monophasic form (usually spindled component only, but occasionally epithelioid only) that usually involves the SSX2 gene
The translocation creates a chimeric protein that has the transactivation domain of SS18(SYT) placed in juxtaposition to a repressor domain of SSX
The resulting aberrant regulation of transcription is thought to contribute to oncogenesis
Other complex chromosomal rearrangements occur in up to two-thirds of synovial sarcomas
♦
RT-PCR using primers that can detect both SSX1 and SSX2 involvement are commonly used. Alternatively, FISH can be used to detect the t(X;18) translocation
♦
Since patients with the SS18(SYT)–SSX2 fusion have a better clinical outcome (~80% survival) than those with the SS18(SYT)–SSX1 variant (~40% survival), it is important when using either PCR or FISH to distinguish between them
Dermatofibrosarcoma Protuberans and Giant Cell Fibroblastoma
♦
t(17;22)(q22;q13) COLIA1–PDGFB
♦
Dermatofibrosarcoma protuberans (DFSP) is a low- to intermediate-grade fibrohistiocytic tumor of adults
Giant cell fibroblastoma is a similar tumor occurring in children
♦
While both tumors are characterized by a t(17;22) translocation , they also commonly have supernumerary ring chromosomes that result from the translocation
The translocation and the ring chromosomes produce a fusion gene consisting of the collagen type I alpha1 gene from chromosome 17 and the platelet-derived growth factor beta-chain gene from chromosome 22
The net result of the fusion is to replace the normally strong negative regulators upstream of the PDGFB gene with the COLIA1 promoter , leading to increased unregulated production of PDGFB
PDGFB is a tyrosine kinase whose activation contributes to oncogenesis
Imatinib, a tyrosine kinase inhibitor, is FDA approved for the treatment of DFSP
♦
RT-PCR can be used to detect the fusion gene, but the heterogeneity in the breakpoint locations in the COLIA1 gene necessitates the use of multiple primer pairs
It is prudent to sequence the amplicon after a successful PCR since the sequence similarity between various COLIA1 exons can give rise to spurious products
Low-Grade Fibromyxoid Sarcoma
♦
t(7;16)(q34;p11) FUS(TLS)–CREB3L2
♦
t(11;16)(p11;p11) FUS(TLS)–CREB3L1
♦
Low-grade fibromyxoid sarcoma is a cytologically bland spindle cell tumor typically occurring in deep soft tissues of young to middle-aged adults
♦
The hallmark translocation, t(7;16)(q34;p11), juxtaposes the 5′ region transcriptional activation domain of FUS(TLS) to the 3′ region leucine zipper motif DNA-binding domain of CREB3L2
The resulting chimeric protein leads to unregulated transcriptional control and oncogenesis
This same translocation is also observed in hyalinizing spindle cell tumor with giant rosettes
♦
A similar variant, t(11;16)(p11;p11), which produces the FUS(TLS)–CREB3L1 fusion protein, occurs in a minority of cases (~5%)
♦
The breakpoints in the FUS(TLS), CREB3L2, and CREB3L1 genes occur in a relatively small region and can be identified by either conventional cytogenetics, FISH, or RT-PCR
Congenital/Infantile Fibrosarcoma
♦
t(12;15)(p13;q25) ETV6–NTRK3
♦
Congenital/infantile fibrosarcoma, also known as juvenile fibrosarcoma, is typically diagnosed in newborns in the soft tissue of the extremities, though it can occur in the head, neck, and trunk as well
♦
The t(12;15)(p13;q25) translocation juxtaposes the ETV6 transcriptional factor to the tyrosine kinase domain of NTRK3, which results in constitutive activation of the tyrosine kinase activity in the chimeric protein
♦
Though the translocation can be detected with conventional cytogenetics, the similar size and banding patterns of the involved segments render diagnosis difficult
FISH and RT-PCR have both shown good sensitivity in detecting the lesion
Immunohistochemistry has also been used to detect overexpression of the NTRK3 protein, though RT-PCR is both more sensitive and specific
It should be noted that the ETV6–NTRK3 fusion gene has also been detected in congenital mesoblastic nephroma and secretory carcinomas of the breast and salivary gland
Myxoid/Round Cell Liposarcoma
♦
t(12;16)(q13;p11) FUS(TLS)–DDIT3(CHOP)
♦
t(12;22)(q13;q12) EWSR1– DDIT3(CHOP)
♦
Most myxoid liposarcomas occur in the deep soft tissue of the extremities, especially the thigh
♦
The large majority of cases harbor the t(12;16)(q13;p11) translocation creating a fusion gene containing FUS(TLS) gene (usually exons 4 and 5 or 4–7) and the DDIT3(CHOP) gene (usually at intron 1)
The resulting chimeric protein includes the transcriptional activation domain of FUS(TLS) and the leucine zipper transcriptional activation domain of DDIT3(CHOP)
This chimeric protein is strongly and constitutively expressed and inhibits differentiation of adipocytes
A minority (5%) of myxoid liposarcomas possess a t(12;22)(q13;q12) translocation fusing the EWSR1 gene to the DDIT3(CHOP) gene with a similar mechanism of action
♦
FISH or RT-PCR is used to detect the gene fusions
Round cell liposarcoma is characterized by the same translocations
Clear Cell Sarcoma
♦
t(12;22)(q13;q12) EWSR1–ATF1
♦
t(2;22)(q33;q12) EWSR1–CREB1
♦
Clear cell sarcoma, also known as melanoma of soft parts, occurs in the deep soft tissues of young adults
♦
The t(12;22)(q13;q12) translocation, found in >90% of cases, fuses the ATF1 gene on chromosome 12 with the EWSR1 gene on chromosome 22
The chimeric protein is a constitutively expressed transcriptional activator of unknown target genes
♦
The t(2;22)(q33;q12) translocation, which fuses the CREB1 gene on chromosome 2 with the EWSR1 gene on chromosome 22, has recently been found in ~5% of cases
This subset of tumors lack melanocytic markers and are found almost exclusively in the GI tract
♦
FISH and RT-PCR have been used to detect the gene fusions
Extraskeletal Myxoid Chondrosarcoma
♦
t(9;22)(q22;q12) EWSR1–NR4A3(CHN)
♦
t(9;17)(q22;q11) TAF2N– NR4A3(CHN)
♦
t(9;15)(q22;q21) TCF12– NR4A3(CHN)
♦
t(3;9)(q11;q22) TFG– NR4A3(CHN)
♦
Most extraskeletal myxoid chondrosarcomas occur in the deep soft tissues of the extremities
♦
The t(9;22)(q22;q12) translocation is present in approximately 75% of cases, with t(9;17)(q22;q11) translocation in 15–20% of cases, while t(9;15)(q22;q21) and t(3;9) rearrangements are found in most of the remaining cases
The predominant rearrangement fuses the EWSR1 gene to the CHN gene forming a chimeric protein that possesses the strong transactivation domain of EWSR1 and a full-length NR4A3(CHN)
The t(9;17) rearrangement fuses the TAF2N gene, which is highly homologous to EWSR1, to NR4A3(CHN)
The t(9;15) translocation fuses the TCF12 gene to the NR4A3(CHN) gene
The t(3;9) translocation fuses the TFG gene to the NR4A3(CHN) gene
All four resulting chimeric proteins are specific to extraskeletal myxoid chondrosarcomas and are thought to promote oncogenesis through the abnormal activation of unknown genes
♦
The four rearrangements can be readily detected with conventional cytogenetics
The t(9;22) translocation is detectable by FISH using an EWSR1 break-apart probe
Molecular techniques, such as RT-PCR, can also be used, with the caveat that primer sets for all four rearrangements would need to be included in the reaction mixes
Alveolar Soft Part Sarcoma
♦
der(17)(X;17)(p11;q25) ASPSCR1(ASPL)–TFE3
♦
Alveolar soft part sarcoma occurs mainly in the head and neck region of children but can also occur in deep soft tissues, especially the thigh, of adults
♦
The hallmark rearrangement is a nonreciprocal der(17) (X;17)(p11;q25) translocation resulting in a ASPSCR1(ASPL)–TFE3 fusion gene
Since the rearrangement is nonreciprocal, there is a concomitant loss of 17q25 sequences telomeric to ASPSCR1(ASPL) and a gain of Xp11 sequences telomeric to TFE3, which may contribute to oncogenesis
The ASPSCR1(ASPL)–TFE3 chimeric protein is constitutively expressed leading to transcriptional deregulation of target genes – e.g., ASPSCR1(ASPL)–TFE3 specifically activates MET signaling making ASPS sensitive to MET inhibitors
♦
Most rearrangements can be detected with conventional cytogenetics, but FISH or RT-PCR may be needed in difficult cases
Interestingly, the ASPSCR1(ASPL)–TFE3 fusion gene is also found in a childhood variant of renal cell carcinoma as a result of a balanced t(X;17)(p11;q25) translocation
IHC for TFE3 is a sensitive marker, but not specific (e.g., TFE3 is also positive in some granular cell tumors)
Inflammatory Myofibroblastic Tumor
♦
ALK (2p23) gene rearrangements
♦
Inflammatory myofibroblastic tumors (IMTs) occur most often in children and young adults and can involve both soft tissues and visceral organs
♦
ALK rearrangements are detected in approximately 50% of IMTs
♦
The ALK gene encodes a membrane-associated tyrosine kinase which is constitutively activated when fused with several partner genes through a variety of chromosomal translocations
♦
Most rearrangements can be detected with immunohistochemistry, but FISH may be needed in difficult cases
♦
There is emerging evidence that ALK-targeted therapies may be an option for the treatment of ALK-positive IMTs
Epithelioid Hemangioendothelioma
♦
t(1;3)(p36;q23-24) WWTR1–CAMTA1
♦
Epithelioid hemangioendotheliomas occur in bone and soft tissues as well as visceral organs, often occurring in the liver and lungs
♦
The hallmark rearrangement is a t(1;3)(p36;q23-24) translocation resulting in a WWTR1–CAMTA1 fusion that places the CAMTA1 transcription factor under the control of the WWTR1 promoter, leading to overexpression of CAMTA1
This translocation is unique to epithelioid hemangioendothelioma (absent in all histologic mimics) and is present in about 85% of cases
Three variants of the WWTR1–CAMTA1 chimeric transcript have been described
♦
This rearrangement can be readily detected by FISH or conventional cytogenetics
Nodular Fasciitis
♦
t(17;22)(p13;q13) MYH9–USP6
♦
Nodular fasciitis is a rapidly growing, benign soft tissue mass that is often reported to arise after mild trauma to the affected area
♦
Recent research suggests this lesion is indeed a clonal lesion that is best categorized as a “transient neoplasia”
♦
Most cases carry the balanced translocation t(17;22)(p13;q13) resulting in MYH9–USP6 gene fusion
The translocation fuses the promoter region of MYH9 to the USP6 gene, resulting in upregulation of the entire coding sequence of USP6, which is a deubiquitinating protease involved in cell trafficking, protein signaling, and inflammation
♦
This rearrangement can be readily detected by FISH or conventional cytogenetics
IDH1 and IDH2 in Chondrogenic Tumors
♦
Recently, mutations in the IDH1 and IDH2 genes have been detected in 50% of solitary enchondromas and 90% of patients with enchondromatosis
♦
IDH1 and IDH2 mutations are also found in 40–70% of primary and ~85% of secondary central chondrosarcomas and in 100% of periosteal chondrosarcomas, although only small studies have been reported thus far
♦
The IDH1 mutations usually occur in residue R132, which is in the active site of the enzyme, and mostly consist of point mutations resulting in an Arg to His amino acid substitution. Similarly, mutations of IDH2 occur in the corresponding codon R172
♦
These mutations confer a neomorphic enzyme activity resulting in the production and accumulation of 2-hydroxyglutarate, an oncometabolite
♦
These mutations are believed to be early events in tumor formation and are not as of yet implicated in malignant transformation
♦
IDH mutations (and the resultant oncometabolite, 2-hydroxyglutarate) are a promising target for several experimental cancer therapies now undergoing clinical trials
Melanoma
RAS/RAF/MEK/ERK Pathway
♦
The MAPK (mitogen-activated protein kinase) pathway (Ras/Raf/Mek/Erk), which is mutated in up to 90% of sporadic melanomas , is a critical pathway in numerous cell types that operates by regulating proliferation and cell survival
♦
Activating mutations may occur in any of the constituents of the pathway, although mutations of these genes appear to be mutually exclusive
♦
RAS missense mutations leading to constitutive activation of GTPase activity have been found in 10–20% of melanomas
NRAS mutations account for the vast majority of RAS mutations in melanomas and have been found in 65–81% of congenital nevi
HRAS mutations are detected in 20% of benign Spitz nevi
♦
BRAF activating point mutations leading to activation of its serine–threonine kinase function (notably V600E) are found in about one-half of melanomas
BRAF mutations are more prevalent in benign acquired melanocytic nevi and in cutaneous melanomas arising from preexisting nevi
♦
Activating mutations in KIT, a tyrosine kinase, are found in a subset of acral melanomas (about 10%) and mucosal melanomas (5–20%) but are present in only about 1% of cutaneous melanomas
♦
Activating mutations in GNAQ or GNA11 are found in 90% of uveal melanomas and are mutually exclusive with each other as well as BRAF, RAS, and KIT mutations
♦
Knowledge of the common disruption of the MAPK pathway has led to the introduction of targeted therapies, such as the FDA-approved BRAF and MEK inhibitors (e.g., vemurafenib, dabrafenib, trametinib, and others)
Mechanisms of acquired resistance to BRAF and MEK inhibitors include activation of alternative signaling pathways as well as reactivating the MAP kinase pathway through alternative means
♦
Approx. 10% of cutaneous melanomas occur in the familial setting, and germline mutations in several genes have been detected: CDKN2A (p16), p14/ARF, CDK4, MITF, and BAP1 (also in uveal melanomas)
Glial Tumors
1p–/19q–
♦
Chromosomal analysis is useful for clarifying the diagnosis of diffuse gliomas
Oligodendrogliomas typically show loss of chromosomes 1p and 19q, unlike astrocytic neoplasms which have intact 1p and 19q
♦
The codeletion of segments of chromosomes 1p and 19q in oligodendrogliomas has been correlated with an enhanced response to chemotherapy and radiotherapy and an improved prognosis
Single chromosome losses, 1p or 19q alone, have not been shown to be prognostic
♦
The missing areas in these deletions are relatively large, and the exact deleted genes responsible for the improved clinical outcome are not currently known
♦
The deletions are now routinely screened for in oligodendrogliomas by either FISH or PCR-based loss of heterozygosity analysis
IDH1 and IDH2
♦
Recently, mutations in the IDH1 gene have been detected in 55–80% of grades 2 and 3 oligodendrogliomas and astrocytomas. Less common are mutations in the IDH2 gene
♦
IDH1 mutations are often found in tumors that also have codeletion of chromosomes 1p and 19q
♦
The IDH1 mutations usually occur in residue R132, which is in the active site of the enzyme, and mostly consist of point mutations resulting in an Arg to His amino acid substitution. Similarly, mutations of IDH2 occur in the corresponding codon R172
♦
These IDH mutations lead to the production of a novel oncometabolite 2-hydroxyglutarate
♦
IDH mutations (and the resultant oncometabolite, 2-hydroxyglutarate) are a promising target for several experimental cancer therapies now undergoing clinical trials
♦
Retrospective studies have shown a favorable prognostic outcome in patients with IDH1 and IDH2 mutations
♦
The impact of IDH mutations in glioma on the response rate to approved chemotherapy drugs (temozolomide), as well as the efficacy of novel targeted therapies directed at IDH mutations in glioma, is currently under investigation
EGFR Amplification
♦
EGFR is the most commonly amplified gene in astrocytic tumors and is useful for distinguishing primary glioblastoma (where EGFR amplification occurs in 40–60%) vs secondary glioblastoma (where EGFR amplification is rarely detected)
♦
EGFR amplification in glioma is associated with resistance to traditional chemotherapy
♦
EGFR amplification does not predict response to anti-EGFR-targeted therapies in glioblastoma (GBM), and nearly all GBMs treated with anti-EGFR agents eventually show progression due to both intrinsic and acquired resistance
MGMT Methylation
♦
MGMT promoter methylation (and resultant underexpression) is present in 45–75% of glioblastomas and is associated with longer survival in glioblastoma patients treated with temozolomide (TMZ)
♦
Recent NCCN guidelines recommend treatment with the alkylating chemotherapy agent, temozolomide, in glioma patients harboring methylated MGMT
♦
MGMT promoter methylation can be detected by a variety of methods, including methylation-specific polymerase chain reaction, methylation-specific multiplex ligation-dependent probe amplification, and pyrosequencing (Fig. 12.2)
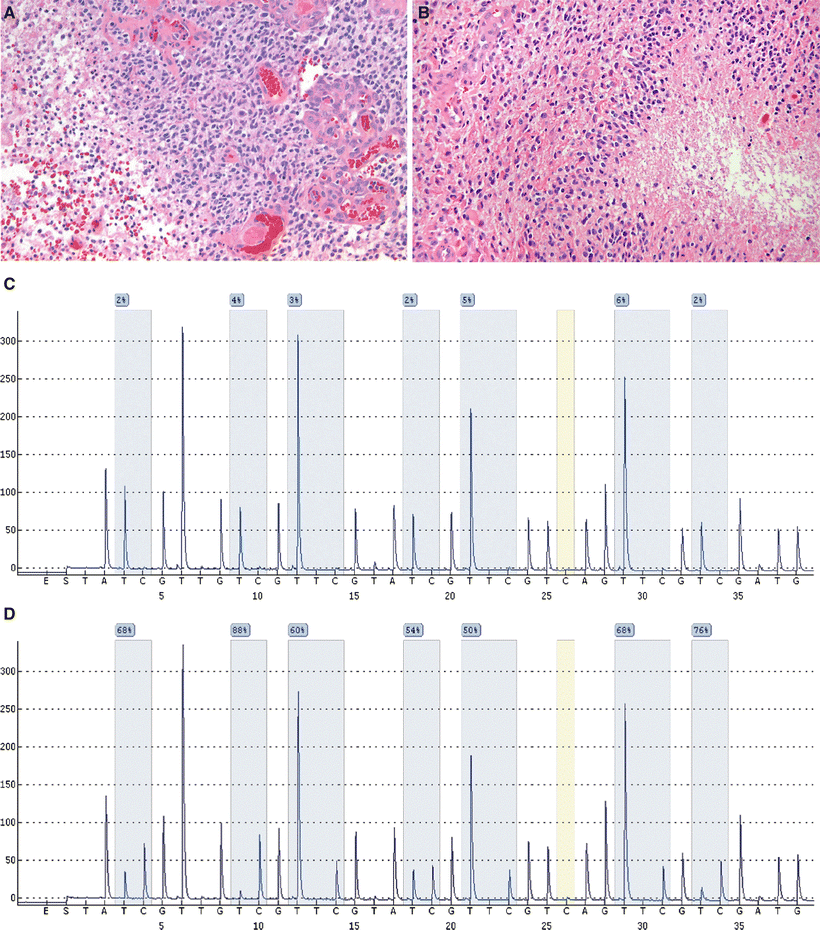
Fig. 12.2.
H&E sections of glioblastoma show marked cellularity with pleomorphic nuclei, prominent endothelial proliferations (A), and pseudopalisading necrosis (B). Pyrograms for MGMT promoter methylation pyrosequencing studies from an unmethylated control (C) and a glioblastoma patient (D). Gray areas indicate CpG sites that are analyzed. Sodium bisulfite treatment of single-stranded DNA selectively deaminates cytosine residues, converting them to uracils, whereas methylcytosine residues remain unaffected. The modified DNA sequence can then be analyzed by a variety of methods (e.g., pyrosequencing, shown here) for evidence of methylation-specific sequence alterations. Methylated cytosine residues are retained as cytosines in the resulting sequence, whereas unmethylated cytosines are converted to uracils and appear as thymidine residues. The degree of methylation is calculated from the peak heights of cytosine and thymidine and each locus, and results are reported as an average of methylation across all interrogated sites.
CpG Island Methylation
♦
Extensive CpG island methylation of genes involving brain development and neuronal differentiation is found in younger patients with low-grade glial tumors and is associated with longer overall survival
Loss of PTEN
♦
Loss of PTEN by IHC is associated with glioma formation and progression, such that low-grade gliomas with loss of PTEN are predicted to behave as glioblastomas
BRAF Mutations and Fusions
♦
BRAF mutations, most commonly the V600E missense mutation, are only occasionally seen in adult gliomas, but are present in about 20% of pediatric high-grade gliomas and up to 85% of pediatric low-grade gliomas
♦
BRAF mutations may suggest responsiveness to BRAF inhibitors in the treatment of glial tumors but may also impact response to anti-EGFR therapies via intrinsic resistance
♦
The BRAF–KIAA1549 gene fusion can be found in 65–70% of pilocytic astrocytomas, particularly those arising in the posterior fossa
Since pilocytic astrocytoma is curable with surgical resection, detecting this fusion may be helpful as a diagnostic marker when histology is indeterminate
Neuroblastoma
MYCN
♦
Neuroblastoma is the most common solid tumor of childhood
As part of the staging process in all new diagnoses, the DNA index and MYCN amplification status are determined by molecular testing
♦
Amplification of the MYCN gene (from 3 to 1,500 copies) on chromosome 2p24 is present in about 25% of neuroblastomas and is associated with an aggressive clinical course
♦
Though MYCN amplification can be detected cytogenetically as double minutes or homogenously staining areas, molecular techniques such as Southern blotting, PCR, or FISH are usually performed
The MYCN gene product is a DNA-binding transcriptional regulator that is important in proliferation and cell survival
♦
Also important in prognostication of neuroblastomas is the determination of the DNA index, which is a measure of ploidy
Diploid tumors have a high proliferative activity and relatively worse outcome, while hyperdiploid tumors tend to be associated with a better prognosis
Diploid tumors are more likely to have MYCN amplification
Medulloblastoma
Molecular Profiling of Medulloblastoma
♦
Based on recommendations from an international consensus conference held in Boston in 2010, medulloblastomas are now stratified into four molecular subgroups (based on gene expression profiling , copy number analysis, and clinical data): these distinct subtypes show different clinical and prognostic features
♦
WNT medulloblastomas, the rarest molecular subtype, are so named for their alterations in the WNT pathway, most of which result in overactivation
The majority show sporadic mutation in CTNNB1 which encodes beta-catenin
WNT medulloblastomas have a very good long-term prognosis with overall survival of approximately 90%
They occur predominantly in children with peak incidence of 10–12 years and are almost completely absent in infants
♦
SHH medulloblastomas, which accounts for 28% of all medulloblastomas, show alterations in the sonic hedgehog pathway which plays a critical role in normal brain development
Sporadic cases commonly show PTCH1 mutations
SHH tumors show an intermediate prognosis between WNT and group 3 tumors
SHH tumors show a bimodal age distribution, being more common in infants less than 4 years and adults more than 16 years
♦
Group 3 medulloblastomas (25% of medulloblastomas) are primarily associated with MYC amplification but rarely MYCN overexpression
Group 3 tumors have the worst overall prognosis, especially when associated with MYCN amplification and a higher rate of metastasis
There is a male predominance among group 3 tumors
♦
Group 4 (34% of medulloblastomas) are associated with isochromosome 17q
These tumors are also associated with MYCN amplification , but minimal MYC overexpression
Group 4 tumors have an intermediate prognosis, and there is a male predominance in this subgroup
♦
The recognition of differences among these four molecular subtypes is changing the treatment strategies for pediatric medulloblastoma
Clinical trials that currently enroll patients with WNT tumors are focusing on reducing the long-term side effects by de-escalating radiation and chemotherapy
The SHH subtype is the first group for which targeted therapies have been used, as clinical trials are employing drugs that target the SHH pathway
Developing molecular-targeted therapies for group 3 and group 4 tumors proposes a considerably greater challenge, as these tumors are affected by pathways that are inherently difficult to target
Currently, there are clinical trials pursuing the use of gemcitabine and pemetrexed for patients with group 3 tumors
Head And Neck Cancer
Squamous Cell Carcinoma
♦
Squamous cell carcinoma of the head and neck (HNSCC) is thought to arise from the multistep accumulation of mutations progressing from dysplasia to invasive carcinoma to metastasis
It is the net accumulation of the mutations and not necessarily the order in which they occur that is important
No single alteration appears to be necessary for any particular stage in tumorigenesis
♦
The “field cancerization theory ” hypothesizes that multiple genetic and epigenetic abnormalities affecting the epithelial surface of the upper aerodigestive tract predispose the region to malignant transformation
This theory suggests a monoclonal origin (e.g., premalignant “field lesion”) to account for the high incidence of second primary tumors (10–35%) within the local region
Genetic and chromosomal alterations are present in HNSCC cells, in their associated premalignant lesions, and in the adjacent histologically benign mucosa
♦
Loss of heterozygosity at multiple loci and numerous gross chromosomal deletions and duplications are prevalent in HNSCC
Dual loss of heterozygosity at 3p and 9p in premalignant conditions predicts progression to invasive carcinoma
♦
Epidermal growth factor receptor (EGFR) overexpression is observed in greater than 90% of HNSCC, indicating a role for anti-EGFR-targeted therapies in HNSCC
Increased EGFR expression correlates with locoregional recurrence and a poorer prognosis
Anti-EGFR-targeted therapies have been approved for treatment of head and neck cancers, but have shown only moderate success as a single therapeutic agent; ongoing research studies are attempting to stratify factors associated with responsiveness and resistance to treatment
♦
Functional alterations in the TP53 tumor suppressor protein are nearly universal in HNSCC
HNSCC with mutated TP53 strongly correlates with a history of tobacco use, a poorer prognosis, and HPV-negative status
♦
HER2 overexpression , although rare in HNSCC, has been reported in association with a worse prognosis, increased recurrence, and decreased overall survival in HNSCC
HER2 has been found to be coexpressed with EGFR in HNSCC tumors and is currently under investigation for its possible role in resistance to anti-EGFR inhibitors
♦
High-risk human papilloma virus subtypes, HPV16 and HPV18 , are the causative agents of 25–35% of HNSCC, more commonly associated with lesions of the oral cavity and oropharynx (~70% of oropharyngeal SCC)
HPV-associated HNSCC accounts for the dramatic shift in HNSCC demographics toward a younger patient population without significant exposure to tobacco and alcohol
HPV-associated lesions tend to be poorly differentiated with increased risk of cervical lymph node involvement but show increased responsiveness to treatment and a better overall survival compared to HPV-negative (often tobacco-related) HNSCC
Immunohistochemistry stains for p16 protein serve as a surrogate marker for HPV infection (National Comprehensive Cancer Network guidelines for head and neck cancer recommend either p16 IHC or HPV DNA testing as a prognostic indicator)
The role of HPV vaccines in HNSCC as a therapeutic and/or preventive measure is currently under investigation
♦
Other genes that are commonly mutated in HNSCC include tumor suppressor genes such as RB1 and p16 and oncogenes such as CCND1 (cyclin D1 gene) and p21
♦
Molecular analysis of HNSCC has demonstrated prognostic value superior to conventional histologic criteria once the mutation profile of a particular tumor is known
Screening for mutations in tissue from surgical margins shows that the risk of tumor recurrence is significantly increased when the mutations are detectable in the surgical margins (i.e., TP53 alterations and allelic loss show sound prognostic value)
HPV and p16 positivity have shown predictive value for lymph node metastasis
For regional lymph node metastases with poor or squamous differentiation and an unknown primary:
HPV positivity is indicative of oral cavity and oropharyngeal tumors
Epstein–Barr virus (EBV) positivity occurs almost exclusively in nasopharyngeal carcinoma
Translocations involving 15q14, most commonly the BRD4–NUT fusion protein resulting from t(15;19)(q14;p13.1), are diagnostic of NUT midline carcinoma, an aggressive lesion of the sinonasal cavity with focal squamous differentiation
The role of multiple mRNA biomarkers in saliva is currently under investigation for potential use for disease screening purposes in high-risk populations
♦
The recent discovery of the role of the programmed death ligand 1 (PD-L1) and its receptor (PD-1) in HNSCC offers a unique opportunity for a new class of molecularly targeted immunotherapies
PD-L1 expression is upregulated in solid tumors where it protects tumor cells from apoptosis and inhibits the immune response by PD-L1:CD80 interactions with cytotoxic CD8 T cells, thereby reducing tumor specific effector immunity
Recent studies have shown high level of PD-L1 expression in human SCCHN tissue, as well as melanoma, lung cancer, and renal cell carcinoma
A new class of monoclonal antibodies (mAbs) has emerged, dubbed as immunotherapy, which are engineered to block or activate specific cosignaling pathways involved in adaptive tumor immunity (such as PD-L1/PD-1), rather than directly targeting the tumor
Anti-PD-1 or anti-PD-L1 mAbs are currently in clinical trials
Salivary Gland Neoplasms
Molecular Biomarkers and Targeted Therapies for Salivary Gland Neoplasms
♦
Recently, many studies have focused on the molecular characterization of salivary gland tumors with the goal of identifying diagnostic and prognostic biomarkers as well as potential molecular-targeted therapies
♦
Several molecular and cytogenetic alterations have been found to be clinically useful with a high degree of specificity in this diagnostic setting (specific examples are described below with their particular diagnostic entities)
♦
Although potential therapeutic targets have been identified in a variety of salivary gland tumors, the response rates observed in most clinical trials have been disappointing
Overexpression of KIT by IHC has been described in the vast majority of adenoid cystic carcinomas (ACC), yet few mutations are identified by molecular analysis; this may account for the lack of response of ACC to agents that target KIT activating mutations
Similar results have been observed in trials for EGFR inhibitors and anti-HER2 agents: the majority of tumors showing overexpression of EGFR and HER2 by IHC do not harbor mutations or gene amplifications, and there has been no significant response to these therapies
Pleomorphic Adenoma and Carcinoma Ex Pleomorphic Adenoma
♦
Pleomorphic adenoma (PA) is the most common benign neoplasm of the salivary glands which typically displays a broad spectrum of histologic features; carcinoma ex pleomorphic adenoma (CXPA) is a malignant salivary gland tumor that arises in association with PA
♦
Alterations involving the pleomorphic adenoma gene 1 (PLAG1 gene), located at chromosomal region 8q12, are present in more than half of PAs and in the majority of CXPAs
In the majority of cases, a t(3;8) translocation results in a CTNNB1–PLAG1 fusion gene
PLAG1 encodes a transcription factor that is also commonly overexpressed in PAs without the t(3;8) translocation
The CTNNB1 gene encodes a beta-catenin protein which plays a role in cell adhesion that is vital for the creation and maintenance of epithelial cell layers
♦
About 10% of PA and CXPAs will show alterations involving 12q15 with overexpression of the HMGA2 gene, which encodes a nonhistone chromatin-associated protein
♦
PLAG1 and HMGA2 gene rearrangements are rare in de novo carcinomas and benign histologic mimics of PA
♦
Conventional cytogenetics, FISH, and PCR-based techniques have all been used to detect the rearrangements involving the PLAG1 and HMGA2 genes
Mucoepidermoid Carcinoma
♦
Mucoepidermoid carcinoma (MEC) is the most common malignant neoplasm of the salivary gland
♦
Regardless of its anatomic site of origin, the hallmark genetic features of MEC are rearrangements involving 11q21 and 19p13, found in 60–80% of MECs
The rearrangements produce a fusion between MECT1 (aka CRTC1) on chromosome 19p13 and the MAML2 gene on 11q21, in which exon 1 of MECT1 is fused to exons 2 through 5 of MAML2
Alterations in both the MECT1 gene (aka CRTC1), which encodes a CREB-regulated transcription coactivator, and the MAML2 gene, a NOTCH1-regulated transcription coactivator, have been found in association with cutaneous mucoepidermoid carcinomas and cancers of the lacrimal gland
The resultant chimeric protein likely acts as an activator of genes regulated by cAMP response elements and genes involved in the Notch pathways, two signaling pathways involved in tumorigenesis
♦
The exact rearrangement, while structurally diverse, has been detected with conventional cytogenetics, FISH, and PCR-based assays, with RT-PCR having the highest sensitivity
A t(11;19) translocation resulting in a MECT1–MAML2 fusion gene has also been detected in a subset of cases of Warthin tumors
Adenoid Cystic Carcinoma
♦
Adenoid cystic carcinoma (ACC) is a well-described salivary gland neoplasm best known for its predilection for extensive perineural invasion and “relentless” clinical course
♦
Recent studies have shown recurrent translocations, t(6;9)(q22-23;p23-24), present in 80–90% of ACC and absent in all other salivary gland neoplasms
The rearrangement produces a fusion between the MYB oncogene on chromosome 6 and the NFIB transcription factor gene on chromosome 9, resulting in increased expression of MYB, a transcription factor which controls cell proliferation and apoptosis
MYB expression is increased, and most ACC show strong nuclear IHC staining for MYB
While finding a drug that specifically targets MYB has proven elusive, clinical trials that target more “druggable” downstream effectors are ongoing
Hyalinizing Clear Cell Carcinoma
♦
Hyalinizing clear cell carcinoma (HCCC) is a malignant epithelial neoplasm comprised of a monomorphic population of cells with optically clear cytoplasm
♦
In the past, HCCC was a diagnosis of exclusion; a newly described recurrent translocation present in about 80% of lesions has improved the diagnostic workup for this entity
The t(12;22)(q13;q12) translocation results in rearrangements involving the EWSR1 gene, most often with fusion to the ATF1 gene, both of which encode transcription factor proteins
While no other salivary gland neoplasm harbors this translocation, it is found in clear cell odontogenic carcinoma which is histologically similar to HCCC, suggesting these entities are closely related, if not identical
Mammary Analogue Secretory Carcinoma
♦
Mammary analogue secretory carcinoma (MASC) is a newly described, though not rare, salivary gland neoplasm named for its histopathologic, immunohistochemical, and molecular features that are almost identical to secretory carcinoma of the breast
♦
The majority of MASCs harbor a balanced translocation t(12;15)(p13; q25), which results in the ETV6–NTRK3 fusion gene, identical to that found in secretory carcinoma of the breast
The chimeric ETV6–NTRK3 (EN) protein joins the SAM-mediated dimerization domain of the ETV6 transcription factor to the protein tyrosine kinase domain of the NTRK3 and activates multiple signaling cascades including Ras–MAP kinase and PI3K–AKT
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


