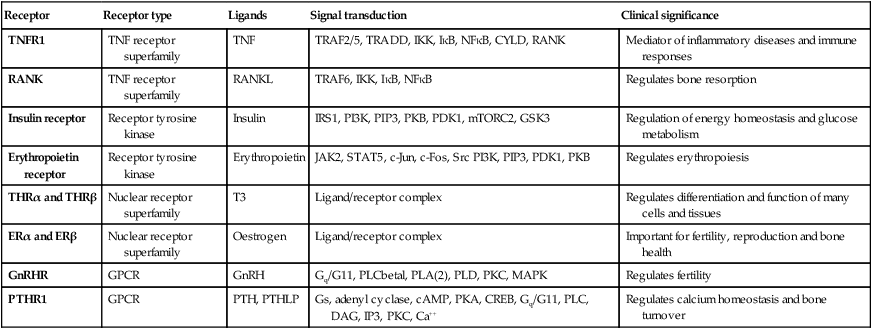
The genome comprises approximately 3.1 billion bp of DNA. Humans are diploid organisms, meaning that each nucleus contains two copies of the genome, visible microscopically as 22 identical chromosomal pairs – the autosomes – named 1 to 22 in descending size order (see Fig. 3.11, p. 57), and two sex chromosomes (XX in females and XY in males). Each DNA strand consists of a linear sequence of four bases – guanine (G), cytosine (C), adenine (A) and thymine (T) – covalently linked by phosphate bonds. The sequence of one strand of double-stranded DNA determines the sequence of the opposite strand because the helix is held together by hydrogen bonds between adenine and thymine or guanine and cytosine nucleotides. Genes are functional elements on the chromosome that are capable of transmitting information from the DNA template via the production of messenger ribonucleic acid (mRNA) to the production of proteins. The human genome contains an estimated 21 500 genes, although many of these are inactive or silenced in different cell types. For example, although the gene for parathyroid hormone (PTH) is present in every cell, activation of gene expression and production of PTH mRNA is virtually restricted to the parathyroid glands. Genes that are active in different cells undergo transcription, which requires binding of an enzyme called RNA polymerase II to a segment of DNA at the start of the gene termed the promoter. Once bound, RNA polymerase II moves along one strand of DNA, producing an RNA molecule that is complementary to the DNA template. A DNA sequence close to the end of the gene, called the polyadenylation signal, acts as a signal for termination of the RNA transcript (Fig. 3.1). The activity of RNA polymerase II is regulated by transcription factors. These proteins bind to specific DNA sequences at the promoter, or to enhancer elements that may be many thousands of base pairs away from the promoter. A loop in the chromosomal DNA brings the enhancer close to the promoter, enabling the bound proteins to interact. The human genome encodes approximately 1200 different transcription factors, and mutations in many of these can cause genetic diseases (Fig. 3.2). Mutation of the transcription factor binding sites within promoters or enhancers also causes genetic disease. For example, the blood disorder alpha-thalassaemia can result from loss of an enhancer located more than 100 000 bp from the alpha-globin gene promoter, leading to greatly reduced transcription. Similarly, variation in the promoter of the gene encoding intestinal lactase determines whether or not this is ‘shut off’ in adulthood, producing lactose intolerance. The accessibility of promoters to RNA polymerase II depends on the structural configuration of chromatin. Transcriptionally active regions have decondensed (or ‘open’) chromatin (euchromatin). Conversely, transcriptionally silent regions are associated with densely packed chromatin called heterochromatin. Chemical modification of both the DNA and core histone proteins allows heterochromatic regions to be distinguished from open chromatin. DNA can be modified by addition of a methyl group to cytosine molecules (methylation). In promoter regions, this silences transcription, since methyl cytosines are usually not available for transcription factor binding or RNA transcription. The core histones can also be modified via methylation, phosphorylation, acetylation or sumoylation at specific amino acid residues in a pattern that reflects the functional state of the chromatin; this is called the histone code – reflecting an emerging understanding of the ‘rules’ by which specific modifications mark transcriptionally activating (trimethylation of lysine 4 on histone H3; acetylation of many histone residues) or silencing (methylation of lysine 9 on histone H4; deacetylation of many histone residues) effects. Such DNA and protein modifications are termed epigenetic, as they do not alter the primary sequence of the DNA code but have biological significance in chromosomal function. Abnormal epigenetic changes are increasingly recognised as important events in the progression of cancer, allowing expression of genes which are normally silenced during development to support cancer cell de-differentiation (see Box 3.3, p. 54). They also afford therapeutic targets. For instance, the histone deacetylase inhibitor vorinostat has been successfully used to treat cutaneous T-cell lymphoma, due to the re-expression of genes that had previously been silenced in the tumour. These genes encode transcription factors which promote T-cell cell differentiation as opposed to proliferation, thereby causing tumour regression. The nascent RNA molecule then undergoes splicing, to generate the shorter, ‘mature’ mRNA molecule, which provides the template for protein production. Splicing removes the regions of the nascent RNA molecule that are not required to make protein (intronic regions), and retains and rejoins those segments that are necessary for protein production (exonic regions). Splicing is a highly regulated process that is carried out by a multimeric protein complex called the spliceosome. Following splicing, the mRNA molecule is exported from the nucleus and used as a template for protein synthesis. It should be noted that many genes produce more than one form of mRNA (and thus protein) by a process termed alternative splicing. Different proteins from the same gene can have entirely distinct functions. For example, in thyroid C cells the calcitonin gene produces mRNA encoding the osteoclast inhibitor calcitonin (p. 738), but in neurons the same gene produces an mRNA with a different complement of exons via alternative splicing, which encodes the neurotransmitter calcitonin-gene-related peptide. Following splicing and export from the nucleus, mRNAs associate with ribosomes, which are the sites of protein production (see Fig. 3.1). Each ribosome consists of two subunits (40S and 60S), which comprise non-coding rRNA molecules complexed with proteins. During translation, tRNA binds to the ribosome. The tRNAs deliver amino acids to the ribosome so that the newly synthesised protein can be assembled in a stepwise fashion. Individual tRNA molecules bind a specific amino acid and ‘read’ the mRNA ORF via an ‘anticodon’ of three nucleotides that is complementary to the codon in mRNA. A proportion of ribosomes are bound to the membrane of the endoplasmic reticulum (ER), a complex tubular structure that surrounds the nucleus. Proteins synthesised on these ribosomes are translocated into the lumen of the ER, where they undergo folding and processing. From here the protein may be transferred to the Golgi apparatus, where it undergoes post-translational modifications, such as glycosylation (covalent attachment of sugar moieties), to form the mature protein that can be exported into the cytoplasm or packaged into vesicles for secretion. The clinical importance of post-translational modification of proteins is shown by the severe developmental, neurological, haemostatic and soft-tissue abnormalities that occur in patients with mutations of the enzymes that catalyse the addition of chains of sugar moieties to proteins. An example is phosphomannose isomerase deficiency, in which there is a defect in the conversion of fructose-6-phosphate to mannose-6-phosphate. This results in a defect in supply of D-mannose derivatives for glycosylation of a variety of proteins, resulting in a multi-system disorder characterised by protein-losing enteropathy, hepatic fibrosis, coagulopathy and hypoglycaemia. Post-translational modifications can also be disrupted by the synthesis of proteins with abnormal amino acid sequences. For example, the most common mutation in cystic fibrosis (ΔF508) results in an abnormal protein that cannot be exported from the ER and Golgi. The mitochondrion is the main site of energy production within the cell. Mitochondria arose during evolution via the symbiotic association with an intracellular bacterium. They have a distinctive structure with functionally distinct inner and outer membranes. Mitochondria produce energy in the form of adenosine triphosphate (ATP). ATP is mostly derived from the metabolism of glucose and fat (Fig. 3.3). Glucose cannot enter mitochondria directly but is first metabolised to pyruvate via glycolysis. Pyruvate is then imported into the mitochondrion and metabolised to acetyl-coenzyme A (CoA). Fatty acids are transported into the mitochondria following conjugation with carnitine and are sequentially catabolised by a process called β-oxidation to produce acetyl-CoA. The acetyl-CoA from both pyruvate and fatty acid oxidation is used in the citric acid (Krebs) cycle – a series of enzymatic reactions that produces CO2, NADH and FADH2. Both NADH and FADH2 then donate electrons to the respiratory chain. Here these electrons are transferred via a complex series of reactions resulting in the formation of a proton gradient across the inner mitochondrial membrane. The gradient is used by an inner mitochondrial membrane protein, ATP synthase, to produce ATP, which is then transported to other parts of the cell. Dephosphorylation of ATP is used to produce the energy required for many cellular processes. Each mitochondrion contains 2–10 copies of a 16 kilobase (kB) double-stranded circular DNA molecule (mtRNA). mtDNA contains 13 protein-coding genes, all involved in the respiratory chain, and the ncRNA genes required for protein synthesis within the mitochondria (see Fig. 3.3). The mutational rate of mtDNA is relatively high due to the lack of protection by chromatin. Several mtDNA diseases characterised by defects in ATP production have been described. mtDNA diseases are inherited exclusively via the maternal line (see Fig. 3.7, p. 51). This unusual inheritance pattern exists because all mtDNA in an individual is derived from that person’s mother via the egg cell, as sperm contribute no mitochondria to the zygote. Mitochondria are most numerous in cells with high metabolic demands, such as muscle, retina and the basal ganglia, and these tissues tend to be the ones most severely affected in mitochondrial diseases (Box 3.1). There are many other mitochondrial diseases that are caused by mutations in nuclear genes, which encode proteins that are then imported into the mitochondrion and are critical for energy production: for example, Leigh’s syndrome and complex I deficiency. 2mtDNA subunits = number of different protein subunits in each complex that are encoded in the nDNA and mtDNA respectively. The cell membrane is a phospholipid bilayer, with hydrophilic surfaces and a hydrophobic core (Fig. 3.4). The cell membrane is, however, much more than a simple wall. Cholesterol-rich ‘rafts’ float within the membrane, and proteins are anchored to them via the post-translational addition of complex lipid moieties. The membrane also hosts a series of transmembrane proteins that function as receptors, pores, ion channels, pumps and associated energy suppliers. These proteins allow the cell to monitor the extracellular milieu, import crucial molecules for function, and exclude or exchange unwanted substances. Many protein–protein interactions within the cell membrane are highly dynamic, and individual peptides will associate and disassociate to effect specific roles. The cell membrane is permeable to hydrophobic substances, such as anaesthetic gases. Water is able to pass through the membrane via a pore formed by aquaporin proteins; mutations of an aquaporin gene cause congenital nephrogenic diabetes insipidus (p. 794). Most other molecules must be actively transported using either channels or pumps. Channels are responsible for the transport of ions and other small molecules across the cell membrane. They open and close in a highly regulated manner. The cystic fibrosis transmembrane conductance regulator (CFTR) is an example of an ion channel that is responsible for transport of chloride ions across epithelial cell membranes. Mutation of the CFTR chloride channel, highly expressed in the lung and gut, leads to defective chloride transport, producing cystic fibrosis. Pumps are highly specific for their substrate and often use energy (ATP) to drive transport against a concentration gradient. Endocytosis is a cellular process that allows internalisation of larger complexes and molecules by invagination of plasma membrane to create intracellular vesicles. This process is typically mediated by specific binding of the particle to surface receptors. An important example is the binding of low-density lipoprotein (LDL) cholesterol-rich particles to the LDL receptor (LDLR) in a specialised region of the membrane called a clathrin pit. In some cases of familial hypercholesterolaemia (p. 453), LDLR mutations cause failure of this binding and thus reduce cellular uptake of LDL. Other LDLR mutations change a specific tyrosine in the intracellular tail of the receptor, preventing LDLR from concentrating in clathrin-coated pits and hence impairing uptake of LDL, even though LDLR bound to LDL is present elsewhere in the cell membrane. The shape and structure of the cell are maintained by the cytoskeleton, which consists of a series of proteins which form microfilaments (actin), microtubules (tubulins) and intermediate filaments (keratins, desmin, vimentin, laminins) that facilitate cellular movement and provide pathways for intracellular transport. Dysfunction of the cytoskeleton may result in a variety of human disorders. For instance, some keratin genes encode intermediate filaments in epithelia. In epidermolysis bullosa simplex (p. 1292), mutations in keratin genes (KRT5, KRT14) lead to cell fragility, producing the characteristic blistering on mild trauma. There are many different signalling pathways; for example, in nuclear steroid hormone signalling, the ligands (steroid hormones or thyroid hormone) bind to their cognate receptor in the cytoplasm of target cells and the receptor/ligand complex then enters the nucleus, where it acts as a transcription factor to regulate the expression of target genes (Box 3.2). However, the most diverse and abundant types of receptor are located at the cell surface, and these activate gene expression and cellular responses indirectly. Activation of a cell surface receptor by its ligand results in a series of intracellular events, involving a cascade of phosphorylation of specific residues in target proteins by an important group of enzymes called kinases. This cascade typically culminates in phosphorylation and activation of transcription factors, which bind DNA and modulate gene expression. Figure 3.5 depicts some of the signalling molecules downstream of the tumour necrosis factor (TNF) receptor. On activation of the receptor by the ligand (in this case, TNF), other molecules, including TNF-receptor-associated proteins (TRAFs), are recruited to the intracellular domain of the receptor. These regulate the activity of a kinase termed IKKγ, which in turn regulates activity of two further kinases termed IKKα and IKKβ. These regulate degradation of an inhibitory protein called IκB, which normally binds to the effector protein NFκB, holding it in the cytoplasm. On receptor activation, a signal is transmitted through TRAFs and the IKK proteins to cause phosphorylation and degradation of IκB, allowing NFκB to translocate to the nucleus and activate gene expression. The system also has negative regulators, including the cylindromatosis (CYLD) enzyme, which regulates the activity of TRAFs by de-ubiquitination. Other transmembrane receptors can be grouped into: • ion channel-linked receptors (glutamate and the nicotinic acetylcholine receptor) • G protein-coupled receptors (GnRH, rhodopsin, olfactory receptors, parathyroid hormone receptor) • receptors with kinase activity (insulin receptor, erythropoietin receptor, growth factor receptors) • receptors which have no kinase activity, but interact with kinases via their intracellular domain when activated by ligand (TNF receptor) (see Figure 3.5 and Box 3.2). In normal tissues, molecules such as hormones, growth factors and cytokines provide the signal to activate the cell cycle, a controlled programme of biochemical events that culminates in cell division. During the first phase, G1, synthesis of the cellular components necessary to complete cell division occurs. In S phase, the cell produces an identical copy of each chromosome – which carries the cell’s genetic information – via a process called DNA replication. The cell then enters G2, when any errors in the replicated DNA are repaired before proceeding to mitosis, in which identical copies of all chromosomes are segregated to the daughter cells. The progression from one phase to the next is tightly controlled by cell cycle checkpoints. For example, the checkpoint between G2 and mitosis ensures that all damaged DNA is repaired prior to segregation of the chromosomes. Failure of these control processes is a crucial driver in the pathogenesis of cancer, as discussed in Chapter 11 (p. 262). With the exception of stem cells, human cells have only a limited capacity for cell division. The Hayflick limit is the number of divisions a cell population can go through in culture before division stops and the cell enters a state known as senescence. This ‘biological clock’ is of great interest in the study of the normal ageing process. Rare human diseases associated with premature ageing, called progeric syndromes, have been very helpful in identifying the importance of DNA repair mechanisms in senescence (p. 168). For example, in Werner syndrome, a DNA helicase (an enzyme that separates the two DNA strands) is mutated, leading to failure of DNA repair and premature ageing. A distinct mechanism of cell death is seen in apoptosis, or programmed cell death. Meiosis is a special form of cell division that only occurs in the post-pubertal testis and the fetal and adult ovary (Fig. 3.6). Meiosis differs from mitosis in two main ways; there are two separate cell divisions and before the first of these there is extensive swapping of genetic material between homologous chromosomes, a process known as recombination. The result of recombination is that each chromosome that a parent passes to his or her offspring is a mix of the chromosomes that the parent inherited from his or her own mother and father. The end products of meiosis are sperm and egg cells (gametes), which contain only 23 chromosomes: one of each homologous pair of autosomes and a sex chromosome. When a sperm cell fertilises the egg, the resulting zygote will thus return to a diploid chromosome complement of 46 chromosomes. The sperm determines the sex of the offspring, since 50% of sperm will carry an X chromosome and 50% a Y chromosome, while each egg cell carries an X chromosome. The individual steps in meiotic cell division are similar in males and females. However, the timing of the cell divisions is very different (see Fig. 3.6). In females, meiosis begins in fetal life but does not complete until after ovulation. A single meiotic cell division can thus take more than 40 years to complete. In males, meiotic division does not begin until puberty and continues throughout life. In the testes, both meiotic divisions are completed in a matter of days. Five modes of genetic disease inheritance are discussed below and illustrated in Figures 3.7 and 3.8.
Molecular and genetic factors in disease
Functional anatomy and physiology
DNA, chromosomes and chromatin
Genes and transcription
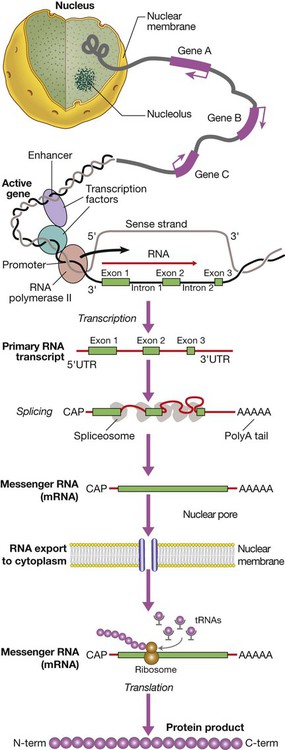
Gene transcription involves binding of RNA polymerase II to the promoter of genes being transcribed with other proteins (transcription factors) that regulate the transcription rate. The primary RNA transcript is a copy of the whole gene and includes both introns and exons, but the introns are removed within the nucleus by splicing and the exons are joined to form the messenger RNA (mRNA). Prior to export from the nucleus, a methylated guanosine nucleotide is added to the 5′ end of the RNA (‘cap’) and a string of adenine nucleotides is added to the 3′ (‘poly A tail’). This protects the RNA from degradation and facilitates transport into the cytoplasm. In the cytoplasm, the mRNA binds to ribosomes and forms a template for protein production.
RNA splicing, editing and degradation
Translation and protein production
Mitochondria and energy production
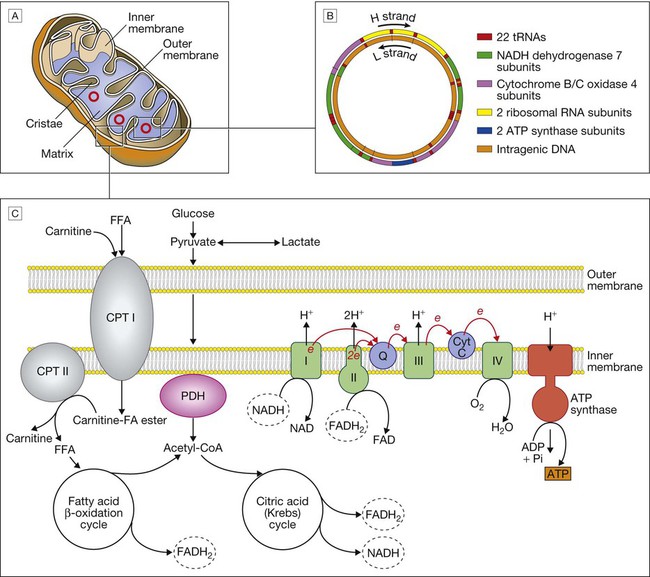
A Mitochondrial structure. There is a smooth outer membrane surrounding a convoluted inner membrane, which has inward projections called cristae. The membranes create two compartments: the inter-membrane compartment, which plays a crucial role in the electron transport chain, and the inner compartment (or matrix), which contains mitochondrial DNA and the enzymes responsible for the citric acid (Krebs) cycle and the fatty acid β-oxidation cycle. B Mitochondrial DNA. The mitochondrion contains several copies of a circular double-stranded DNA molecule, which has a non-coding region, and a coding region which encodes the genes responsible for energy production, mitochondrial tRNA molecules and mitochondrial rRNA molecules. ATP = adenosine triphosphate; NADH = nicotinamide adenine dinucleotide. C Mitochondrial energy production. Fatty acids enter the mitochondrion conjugated to carnitine by carnitine-palmityl transferase type 1 (CPT I) and, once inside the matrix, are unconjugated by CPT II to release free fatty acids (FFA). These are broken down by the β-oxidation cycle to produce acetyl-CoA. Pyruvate can enter the mitochondrion directly and is metabolised by pyruvate dehydrogenase (PDH) to produce acetyl-CoA. The acetyl-CoA enters the Krebs cycle, leading to the production of NADH and flavine adenine dinucleotide (reduced form) (FADH2), which are used by proteins in the electron transport chain to generate a hydrogen ion gradient across the inter-membrane compartment. Reduction of NADH and FADH2 by proteins I and II respectively releases electrons (e), and the energy released is used to pump protons into the inter-membrane compartment. As these electrons are exchanged between proteins in the chain, more protons are pumped across the membrane, until the electrons reach complex IV (cytochrome oxidase), which uses the energy to reduce oxygen to water. The hydrogen ion gradient is used to produce ATP by the enzyme ATP synthase, which consists of a proton channel and catalytic sites for the synthesis of ATP from ADP. When the channel opens, hydrogen ions enter the matrix down the concentration gradient, and energy is released that is used to make ATP.
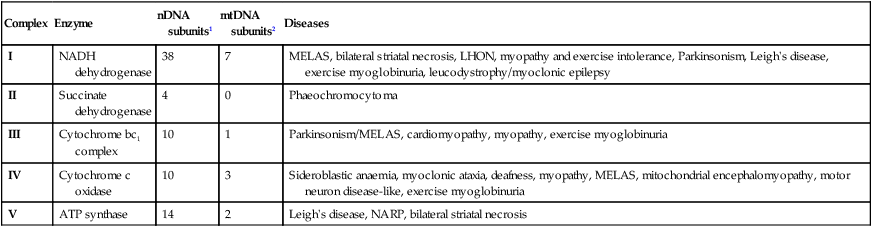
 3.1 The structure of the respiratory chain complexes and the diseases associated with their dysfunction
3.1 The structure of the respiratory chain complexes and the diseases associated with their dysfunction
Complex
Enzyme
nDNA subunits1
mtDNA subunits2
Diseases
I
NADH dehydrogenase
38
7
MELAS, bilateral striatal necrosis, LHON, myopathy and exercise intolerance, Parkinsonism, Leigh’s disease, exercise myoglobinuria, leucodystrophy/myoclonic epilepsy
II
Succinate dehydrogenase
4
0
Phaeochromocytoma
III
Cytochrome bc1 complex
10
1
Parkinsonism/MELAS, cardiomyopathy, myopathy, exercise myoglobinuria
IV
Cytochrome c oxidase
10
3
Sideroblastic anaemia, myoclonic ataxia, deafness, myopathy, MELAS, mitochondrial encephalomyopathy, motor neuron disease-like, exercise myoglobinuria
V
ATP synthase
14
2
Leigh’s disease, NARP, bilateral striatal necrosis
The cell membrane and cytoskeleton
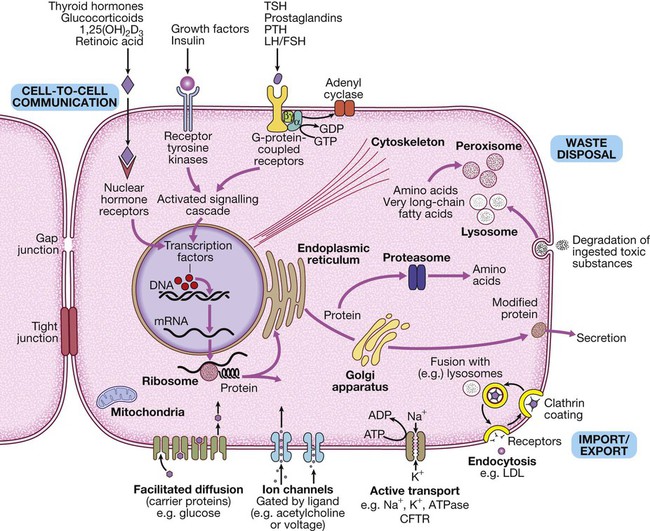
The basic cell components required for function within a tissue: (1) cell-to-cell communication taking place via gap junctions and the various types of receptor that receive signals from the extracellular environment and transduce these into intracellular messengers; (2) the nucleus containing the chromosomal DNA; (3) intracellular organelles, including the mechanisms for proteins and lipid catabolism; (4) the cellular mechanisms for import and export of molecules across the cell membrane. (ABC = ATP-binding cassette transporters; ATP = adenosine triphosphate: cAMP = cyclic adenosine monophosphate; CFTR = cystic fibrosis transmembrane regulator; CREB = cAMP response element-binding protein; GDP/GTP = guanine diphosphate/triphosphate; LDL = low-density lipoproteins; LH/FSH = luteinising hormone/follicle-stimulating hormone; PTH = parathyroid hormone; TSH = thyroid-stimulating hormone)
Receptors, cellular communication and intracellular signalling
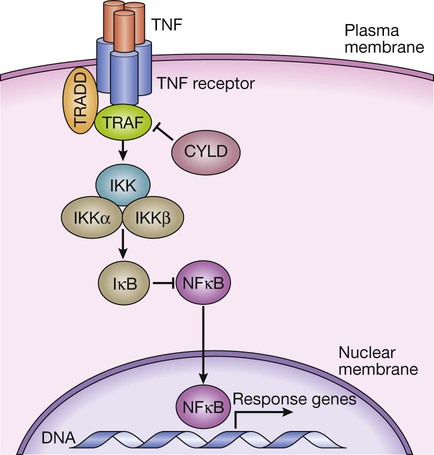
TNF binds to its receptor, forming a trimeric complex in the cell membrane. Various receptor-associated factors are attracted to the intracellular domain of the receptor, including TNF-receptor-associated protein 6 (TRAF6) and tumour necrosis factor receptor type 1-associated death domain protein (TRADD). These proteins modulate activity of downstream signalling proteins, the most important of which are IKKγ (which in turn modulates activity of IKKα and IKKβ). These proteins cause phosphorylation of IκB, which is targeted for degradation by the proteasome, releasing NFκB, which translocates to the nucleus to activate gene expression. The signalling pathway is further regulated in a negative manner by cylindromatosis (CYLD), which de-ubiquitinates TRAF6, thereby impairing its ability to activate downstream signalling.
Cell division, differentiation and migration
Cell death, apoptosis and senescence
Genetic disease and inheritance
Meiosis
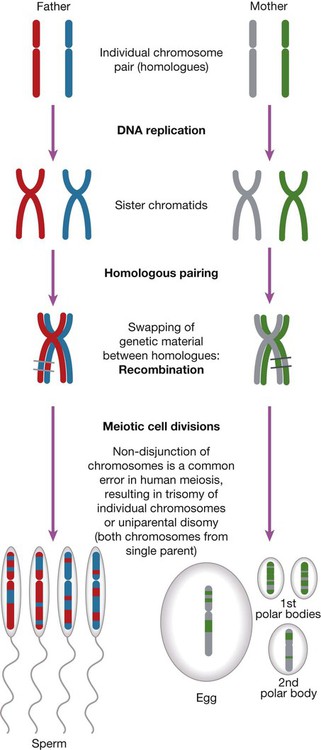
The main chromosomal stages of meiosis in both males and females. A single homologous pair of chromosomes is represented in different colours. The final step is the production of haploid germ cells. Each round of meiosis in the male results in four sperm cells; in the female, however, only one egg cell is produced, as the other divisions are sequestered on the periphery of the mature egg as peripheral polar bodies.
Patterns of disease inheritance
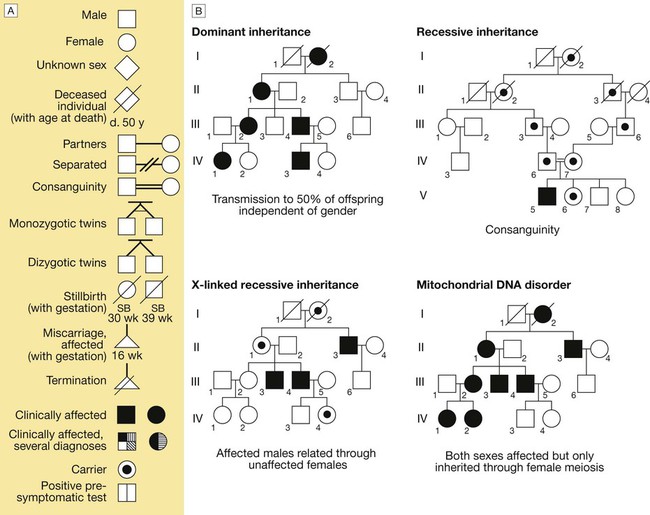
A The main symbols used to represent pedigrees in diagrammatic form. B The main modes of disease inheritance (see text for details).
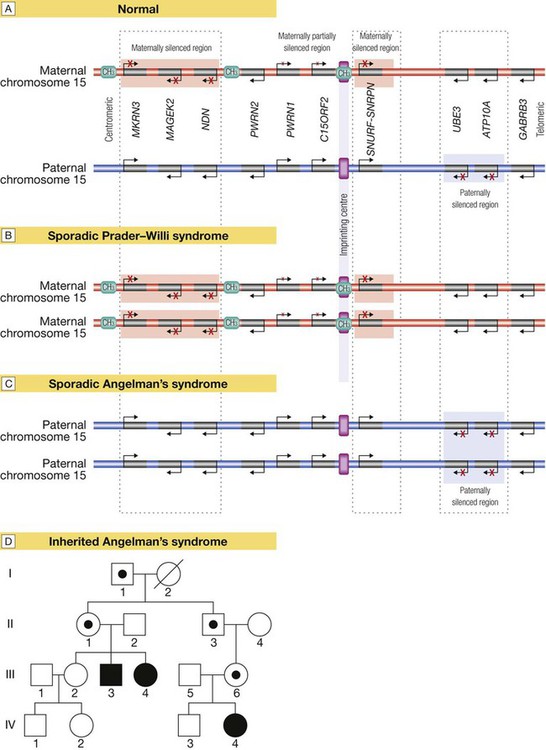
Several regions of the genome exhibit the phenomenon of imprinting, whereby expression of one or a group of genes is influenced by whether the chromosome is derived from the mother or the father; one such region lies on chromosome 15. A Normal imprinting. Under normal circumstances, expression of several genes is suppressed (silenced) on the maternal chromosome (red), whereas these genes are expressed normally by the paternal chromosome (blue). However, two genes in the paternal chromosome (UBE3 and ATP10A) are silenced. B In sporadic Prader–Willi syndrome, there is a non-disjunction defect on chromosome 15, and both copies of the chromosomal region are derived from the mother (maternal uniparental disomy). In this case, Prader–Willi syndrome occurs because there is loss of function of several paternally expressed genes, including MKRN3, MAGEK2, NDN, PWRN2, C15ORF2 and SNURF-SNRNP. C In sporadic Angelman’s syndrome, both chromosomal regions are derived from the father (paternal uniparental disomy) due to non-disjunction during paternal meiosis. As a result, both copies of the UBE3 gene are silenced and this causes Angelman’s syndrome. Note that the syndrome can also be caused by deletion of this region on the maternal chromosome or a loss-of-function mutation on the maternal copy of UBE3, causing an inherited form of Angelman’s, as illustrated in panel D. D Pedigree of a family with inherited Angelman’s syndrome due to a loss-of-function mutation in UBE3. Inheriting this mutation from a father causes no disease (because the gene is normally silenced in the paternal chromosome) (see individuals I-1, II-1, II-3, III-6), but the same mutation inherited from the mother causes the syndrome (individuals III-3, III-4, IV-4), as this is the only copy expressed and the UBE3 gene is mutated.
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Molecular and genetic factors in disease
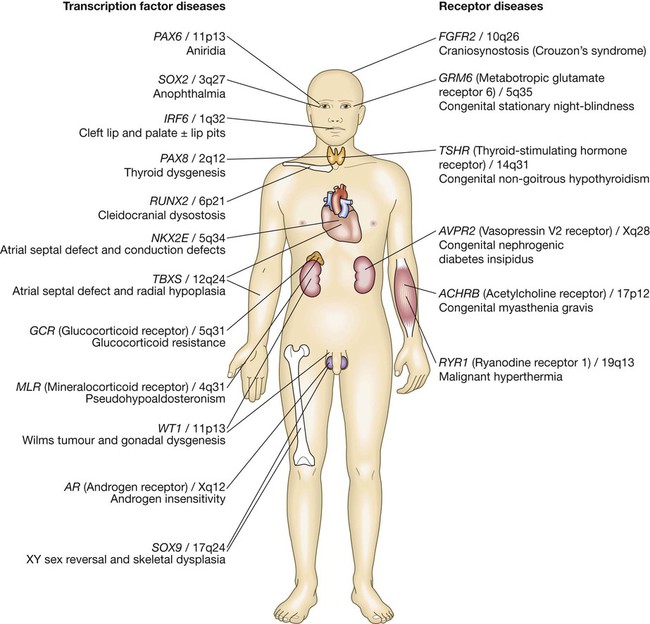
 3.2
3.2