Metastatic Neuroblastoma
Definition
Neuroblastoma metastatic to lymph nodes.
Epidemiology
Neuroblastoma (including ganglioneuroblastoma, discussed later) accounts for 7% to 10% of childhood cancers and is the most common solid tumor of infants. The prevalence of neuroblastoma is approximately 1 case per 7,000 live births, and about 700 new cases occur each year in the United States (1). About 90% of children with neuroblastomas are diagnosed within the first 5 years of life (2). Neuroblastoma is rare in older children and adults. Prevalence does not appear to be related to sex or geographic distribution. Both breast-feeding and dietary fortification with folic acid have been correlated with a lower incidence of neuroblastoma (3,4).
Pathogenesis
The etiology and pathogenesis of neuroblastoma are largely unknown. A genetic predisposition is likely in a subset of cases (5). Patients with hereditary neuroblastoma usually have multiple, bilateral, and multifocal tumors that arise at an early age, with a median age of approximately 9 months. Chromosome 16p12–13 has been mapped as one locus responsible for a hereditary predisposition to neuroblastoma (6). In the literature, hereditary neuroblastoma has been linked with Beckwith-Wiedemann syndrome, neurofibromatosis type 1 (von Recklinghausen disease), Hirschsprung disease, and other more rare congenital abnormalities (7,8,9). However, in one review it was concluded that the association between neuroblastoma and neurofibromatosis type 1 could be accounted for by chance alone (8), and no genetic linkage between neuroblastoma and Hirschsprung disease has been shown (10). Neuroblastoma also has been linked with increased risk of subsequent renal cell carcinoma. The genetic basis for this relationship is unknown (11). Sporadic neuroblastoma is much more common. Sporadic tumors are characteristically solitary and arise in older children, with a median age of 21 months.
Embryonically, neuroblastoma is derived from primitive cells of the sympathetic nervous system, which is part of the neural crest. Thus, neuroblastoma has been included as a part of the various benign and malignant malformations of the neural crest known as neurocristopathies (12). Neuroblastoma can exhibit variable neural maturation, and these tumors represent the undifferentiated end of a spectrum, with fully differentiated and benign ganglioneuroma representing the opposite end of this spectrum (Figs. 96.1 and 96.2) (13). The presence and degree of mature or immature ganglion cells within neuroblastoma directly affects the prognosis (14,15). Ganglioneuroblastoma is a neoplasm that exhibits maturation intermediate between neuroblastoma and ganglioneuroma. Two histologic types are recognized. In one type, the neoplasm has evidence of all stages of neural differentiation uniformly distributed throughout the entire tumor. The second type of ganglioneuroblastoma has two components, with one component being ganglioneuroma and the other component neuroblastoma.
Clinical Syndrome
As expected from its cell of origin, neuroblastomas arise in sites corresponding to the distribution of the sympathetic nervous system (13). In 50% to 75% of cases, they originate in the adrenal medulla. Extra-adrenal neuroblastomas arise in the autonomic ganglia in the orbit, pterygopalatine fossa, cervical region, posterior mediastinum, and paravertebral retroperitoneal space. Disease distribution, to some extent, is determined by age. In the first year of life, almost all neuroblastomas arise in the adrenal glands, whereas in older patients, the tumors are more often extra-adrenal (5).
The most common symptoms in patients with neuroblastoma are pain (34%), fever (28%), and weight loss (21%) (2). Uncommon, but distinctive symptoms occur in neuroblastoma patients as a result of tumor location or products secreted by the neoplasm. These include lower limb paresis, as a result on intraspinal extension of a paraspinal neoplasm; Horner syndrome, with tumors arising in the neck and thorax; severe diarrhea, as a result of the production of vasoactive intestinal peptide; hypertension, as a result of catecholamine secretion; and acute cerebellar encephalopathy, as a result of unknown metabolic disturbances (2). In some patients, the tumors may reach large proportions before causing symptoms.
Prognosis in neuroblastoma correlates, in large part, with patient age and stage. Children younger than 1 year of age have a better prognosis (2). Age is important because a relatively high percentage (∼10%) of neuroblastomas undergo spontaneous regression, and this is more likely in infants. The frequency of spontaneous regression has been estimated to be 10 to 100 times more likely in neuroblastoma than in any other cancer (2). The age of patients is also a factor in the distribution of metastases. Bone marrow and liver metastases are more common before 1 year of age, and metastases to the regional lymph nodes and bones are more frequent in older children (2).
The currently used International Neuroblastoma Staging System is shown in Table 96.1. Patients with low-stage disease are at low risk for metastases, especially if younger than 1 year of age, and these children often do not require chemotherapy. Patients with high-stage disease are considered high-risk and, therefore, require aggressive chemotherapy. In addition, a subset of patients with stage 4 disease have a much better prognosis than other stage 4 patients. These patients are designated as having stage 4S disease (S for special). Stage 4S is defined as distant metastases in the liver, skin, and/or bone marrow without radiologic or other evidence of bone metastases. Stage 4S represents approximately 10% of all stage 4 cases and tends to be more common in infants. These tumors respond well to therapy and often show evidence of spontaneous regression.
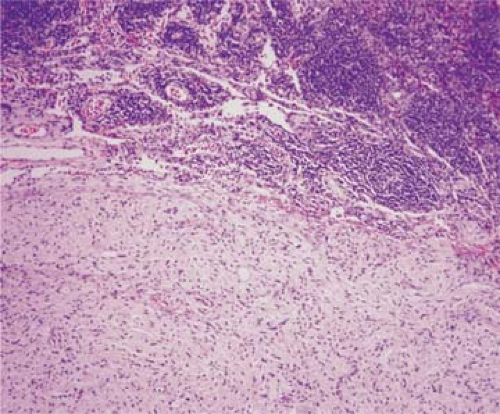 Figure 96.1. Lymph node metastasis of ganglioneuroma. The lymph node (upper part of field) is extensively replaced by the neoplasm. Hematoxylin-eosin stain. |
In general, the prognosis of extra-adrenal neuroblastomas is better than that of tumors originating in the adrenal gland. This may be related to the differentiation of the neoplasm, as adrenal gland neoplasms tend to be poorly differentiated. The presence of lymph node metastases at time of initial diagnosis does not substantially affect overall prognosis (16). Metastases are common, and as many as one-third of patients initially present with metastatic disease. Neuroblastomas metastasize in descending order of frequency to the bone marrow, lymph nodes, liver, skin, orbit, and bones (13). Metastases usually occur within the first 2 years after diagnosis.
Neuroblastoma occurs rarely in adults, and small cases series of adults with neuroblastoma have been reported. Mackay and colleagues (17), in one of the larger series, described nine cases of neuroblastoma in patients ages 18 to 72 years. All arose at extra-adrenal sites, and seven presented at a peripheral location. Hasegawa and colleagues (18) have reported eight cases in the retroperitoneum and abdomen in adults ages 22 to 74 years. Metastases to regional lymph nodes are common.
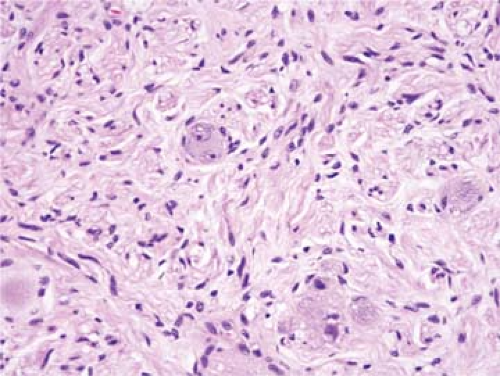 Figure 96.2. Lymph node metastasis of ganglioneuroma. Same case as shown in Fig. 96.1. The neoplasm is composed of Schwann cells and ganglion cells. Hematoxylin-eosin stain. |
TABLE 96.1 INTERNATIONAL NEUROBLASTOMA STAGING SYSTEM | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Histopathology
Lymph node metastases resemble the primary neoplasm (Fig. 96.3). Grossly, the primary neuroblastoma mass is pale gray, with frequent areas of necrosis, hemorrhage, and cyst formation. It may be a unicentric mass or an aggregation of
large tumor nodules. Histologically, neuroblastoma is diffuse or vaguely nodular, the latter being the result of the presence of thin fibrous septa (Fig. 96.4) (13,14,15). Necrosis and hemorrhage are almost always present, and calcification is common, occurring in approximately half of cases. Neuroblastoma is composed of sheets of closely packed, uniform, basophilic tumor cells with indistinct cytoplasm and round, basophilic nuclei that have stippled chromatin (Fig. 96.5). Apoptotic and pyknotic nuclei are frequent. Nuclear pleomorphism is minimal, with only small variations in size and occasional slight indentations. Mitoses are present but are usually not numerous. These are features of the least differentiated neuroblastomas and, without ancillary methods (e.g., immunohistochemistry), the diagnosis of neuroblastoma can only be suggested. In most neuroblastomas, however, some features of differentiation may be present, such as cytoplasmic processes or neurites with a characteristic eosinophilic fibrillary background. In approximately one-third of cases, the neoplastic cells are arranged in Homer-Wright rosettes, in which the nuclei form a circle surrounding a central area occupied by cell processes without a central lumen (Fig. 96.6). In addition, acellular fibrillar areas are seen that resemble glial tissue (neuropil) and become prominent in more differentiated neuroblastomas. Glycogen is usually not present in the cytoplasm of neuroblastoma cells (in contrast with Ewing sarcoma). Other histologic features include precipitation of DNA around blood vessels (the so-called Azzopardi effect) and variable lymphocytic infiltration (13,15). After therapy, neuroblastoma can show evidence of maturation to ganglioneuroma, usually partial.
large tumor nodules. Histologically, neuroblastoma is diffuse or vaguely nodular, the latter being the result of the presence of thin fibrous septa (Fig. 96.4) (13,14,15). Necrosis and hemorrhage are almost always present, and calcification is common, occurring in approximately half of cases. Neuroblastoma is composed of sheets of closely packed, uniform, basophilic tumor cells with indistinct cytoplasm and round, basophilic nuclei that have stippled chromatin (Fig. 96.5). Apoptotic and pyknotic nuclei are frequent. Nuclear pleomorphism is minimal, with only small variations in size and occasional slight indentations. Mitoses are present but are usually not numerous. These are features of the least differentiated neuroblastomas and, without ancillary methods (e.g., immunohistochemistry), the diagnosis of neuroblastoma can only be suggested. In most neuroblastomas, however, some features of differentiation may be present, such as cytoplasmic processes or neurites with a characteristic eosinophilic fibrillary background. In approximately one-third of cases, the neoplastic cells are arranged in Homer-Wright rosettes, in which the nuclei form a circle surrounding a central area occupied by cell processes without a central lumen (Fig. 96.6). In addition, acellular fibrillar areas are seen that resemble glial tissue (neuropil) and become prominent in more differentiated neuroblastomas. Glycogen is usually not present in the cytoplasm of neuroblastoma cells (in contrast with Ewing sarcoma). Other histologic features include precipitation of DNA around blood vessels (the so-called Azzopardi effect) and variable lymphocytic infiltration (13,15). After therapy, neuroblastoma can show evidence of maturation to ganglioneuroma, usually partial.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


