Fig. 7.1
Steps in metagenomic analysis
.
7.2 Steps in Metagenomic Analysis
7.2.1 DNA Isolation
In general, metagenomic DNA isolation strategy should result in representation from a broad range of organisms. DNA isolation is, therefore, the most important step for the subsequent downstream genome analysis. Shearing of DNA should be avoided during extraction as long intact fragments are required for community analysis. Further, the DNA should be free from contaminating elements for easy downstream studies. It is difficult to develop general protocol for DNA isolation as the environmental sample may contain enormous group of microorganisms belonging to different genotypes, classes, and divisions having marked difference in their cell wall structure. In addition to that, problem for recovery of DNA straight from environmental sample lies in the fact that cells are in a physiological state that is hard to lyse. Designing of a unique ideal DNA isolation protocol for recovery from every microbial cell in the sample is, therefore, extremely difficult. Despite every effort certain groups are represented more in terms of the isolated genomic material compared to others. However, in certain cases a genomic biasness is deliberately introduced where the aim is to collect DNA from certain group of bacteria with some desirable properties. There are two main strategies for extraction of metagenomic DNA viz. ‘direct method’ involving in situ cell lysis and ‘indirect method’ where cells are first separated from the sample and then lysed. Both methods are equally good as far as the DNA analysis for determination of diversity spectrum is concerned. However, the former method results in more amount of DNA in an impure form. In case of soil the problem of contaminating agents such as humic acid may interfere with the process of DNA isolation. On the other hand, a large amount of water has to be processed in order to have a significant yield of DNA from aquatic samples.
Vigorous cell lysis methods are generally employed considering the equal representation of each community member from the diverse population in an environmental sample. This may, however, lead to the shearing of genomic fragments and thus making the intact gene recovery difficult. Also, there is an increased risk of chimera formation from small template DNA during subsequent PCR. Extraction of total metagenomic DNA is, therefore, a compromise between the vigorous extraction and quality of DNA required for the representation of all microbial genomes. Other physical methods for DNA isolation include freeze-thawing, ultrasonication, etc. Chemical methods, on the other hand, are preferred when the aim is to select certain taxa by exploiting their unique biochemical properties. SDS-based cell lysis is the most common type of method for release of intracellular DNA. Mechanical method is shown to recover more diversity than the chemical method. A combination of different physical and chemical methods can also be used depending on the soil characteristics and associated microbial diversity.
Since not all members of a community are evenly represented in the extracted genome, experimental normalization is sometimes necessary as a partial solution to it. One such approach involves density gradient separation of genotypes in the presence of an intercalating agent. This leads to the separation of genomes based on their % G and C content as distinct bands in centrifugation tube. A normalized metagenome is then obtained by combining equal amount of each band. Another method for normalization for rare sequences involve denaturing metagenomic DNA followed by re-annealing under stringent conditions. Compared to rare sequences, abundant single-stranded DNA will anneal rapidly and acquire double-stranded form. Single-stranded sequences can then be separated from the double-stranded nucleic acids.
A precultivation step improves the quality of the community DNA. This, of course, reduces the chance of representation of maximum diversity of the original environmental sample and enhances the possibilities of recovering genomes from microorganisms with desired trait. The important strategies that have been employed for enriching the metagenome for sequences of interest before cloning, are summarized in Table 7.1. Other latest methods for enrichment are based on microarray, suppressive subtractive hybridization, differential expression analysis, and multiple displacement amplification.
Table 7.1
Metagenomic library enrichment methods
Enrichment method | Principle | References |
|---|---|---|
Based on GC content | Enrichment for the sequences of interest by its separation from the bulk metagenomic DNA based on the variation in GC contents through ultracentrifugation. | Schloss and Handelsman (2005) |
BrdU enrichment | Incorporation of bromodeoxyuridine (BrdU) in the soil leading to its incorporation into the DNA of metabolically active subset of the community. The labeled DNA can then be separated by immunocapture technique | Urbach et al. (1999) |
Stable isotope probing (SIP) | C13 labeled substrate is provided to the soil bacteria and those capable of metabolizing the substrate incorporate the isotope into their DNA. The heavier DNA due to C13 is separated from the bulk by density gradient centrifugation | Radajewski and Murrell (2002) |
Culture enrichment | The sample can be enriched for desired microbes by varying nutritional, physical, and chemical conditions during precultivation step | Singh et al. (2009) |
7.2.2 Library Construction and Choice of Vector
Metagenomic library has been created in almost all types of commonly used gene cloning vectors such as plasmid, phage, cosmid, fosmid, bacterial artificial chromosome (BAC), etc. During the early studies plasmid-based vectors were primarily used for metagenomic library with less than 10 kb insert size. Large capacity vectors are now fast replacing the plasmid-based vectors. The choice, however, depends on the overall aim of the study. Small insert library is suitable for cloning of individual gene. These libraries are useful for functional screening of clones expressing various enzymes like chitinase, lipase, esterase, amylase, nitrilase, etc. It also provides large amount of sequencing information. The main drawback with small capacity vectors is the huge number of clones in the library which are sometimes very difficult to handle. On the other hand, large insert library is required when gene cluster or operons are to be isolated and studied. Cosmid or fosmid vectors have been used to create library with an insert size of about 40 kb, whereas BAC vectors can markedly reduce the size of the metgenomic library by cloning up to 150 kb. The recovery of high molecular weight DNA is, however, a prerequisite for using the high capacity vectors, which is sometimes very difficult to achieve as the soil compositions demand vigorous isolation strategies leading to extensive fragmentation of DNA. Escherichia coli is the most preferred choice of host. Other hosts such as Streptomyces lividans, Pseudomonas putida, and Rhizobium sp. have also been successfully used.
7.2.3 Screening Strategies
7.2.3.1 Function Based
In a metagenomic library clones with useful traits can be traced by mainly two methods viz. function-based screening and sequence-based screening. The functional screening depends on the faithful expression of the cloned gene in a heterologous host. Since no pre-sequence analysis of the gene of interest is required, there is very high chance to identify entirely new classes of genes for new or known functions. The expression of the gene may confer some selective advantage to the desired clone making its visual identification possible in a pool of clones. For example, antibiotic resistance genes or enzyme producing genes can be selected on specific plates (Fig. 7.2). However, very few clones in a metagenomic library express any given activity. Many thousand clones need to be screened before getting one with desirable trait. In the recent years highly efficient screening and selection systems have been developed for selecting positive clones. Uchiyama et al. (2005) have developed Substrate-induced gene expression screening (SIGEX) for rapid identification of clones showing catabolic gene expression. The technique is based on the fact that catabolite-gene expression requires certain substrates as inducers which, in many cases, is controlled by regulatable elements situated near to catabolic genes. A plasmid-based operon-trap expression vector (p18GFP) containing gene for green fluorescent protein (GFP) immediately after the insert site, is constructed and used for creating metagenomic library in this technique. The clones are exposed to an inducer which easily diffuses to the cyptoplasm of all transformed cells in the library. In positive clones (in which the entire operon responding to that substrate/inducer is cloned), the inducer binds to the cloned regulatory elements and causes expression of downstream sequences for GFP. The GFP positive cells can then be separated by fluorescence activated cell sorter (FACS). Another similar approach is METREX in which product of metagenomic DNA induces bacterial quorum sensing. The metagenomic library is created in host cells which already contain a reporter plasmid. The reporter plasmid contains a reporter gene such as gfp under the control of a regulatory promoter. The promoter can be upregulated in the presence of the product whose gene is present only in the positive clones in the library. The positive clones thus express gfp and can be identified and separated from other clones in the library.
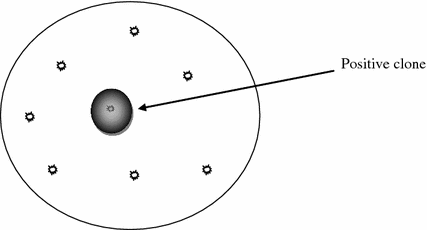
Fig. 7.2
Function-based visual identification of positive clone on solid medium
7.2.3.2 Sequence Based
This strategy is based on the prior information of target gene sequence. The classical approach of hybridization with labeled probe can be used to identify the positive clones in the metagenomic library (Fig. 7.3). Complete sequencing of clones that contains phylogenetic anchors indicates the taxonomic group of the source of DNA. Random sequencing can also be done to identify a gene of interest followed by assessment of flanking DNA to contain any phylogenetic anchor. The powerful approach of sequence analysis guided by the identification of phylogenetic markers produced the first genomic sequence linked to a 16S rRNA gene that affiliated with γ–Proteobacteria (Handelsman 2004). Sequencing random clones is an alternative to phylogenetic marker-based approach. The field of metagenomics has been transformed by the application of a whole genome shotgun sequencing approach during the last 6–7 years. Next-generation ultra high-throughput sequencing techniques have revolutionized the field with heavy price cut allowing sequencing at scale larger then anytime before. Sequencing projects have been carried out to assess the actual microbial diversity and their ecological inference in different environment such as sea and acid mine drainage, etc. In the past few years, techniques of DNA microarray has also been widely used for screening clones in metagenomic library.
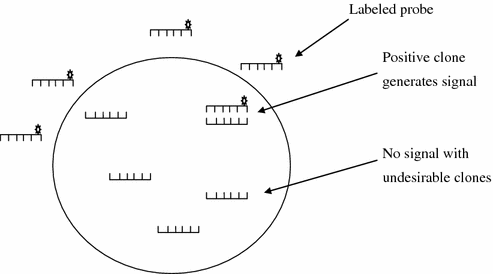
Fig. 7.3
Sequence-based identification of positive clone
The technique of PCR has been applied to the isolation and detection of novel genes from community genome. However, the major limitation with PCR is to rely on the flanking DNA sequence information for isolation of an unknown gene from an uncultured organism. Furthermore, when PCR is directed at conserved sequence motifs, only partial genes are recovered. In an attempt to solve this problem, a novel strategy for recovering complete open reading frame from environmental DNA samples was proposed by Stokes in 2001. His team designed PCR assays targeted toward the 59-base element family of recombination sites that flank integrons associated gene cassettes. This approach has resulted in the amplification of diverse gene cassettes containing complete open reading frame from the environmental DNA.
7.3 Applications of Metagenomics
Metagenomic studies are important and have a range of applications for both academia and industries. The recent findings in metagenomics suggest the role of microorganism in influencing human life far beyond the previous expectations. The important applications and outcome from these studies are summarized below.
7.3.1 Ecological Inferences from Microbial Diversity Estimation
In microbial ecology it is important to know how microorganisms acquire nutrients and produce energy, form symbioses and compete, and communicate with other members of the community. Metagenomic investigations have resulted in the identification of various novel life forms in geographically distinct region and attempts are still underway to describe their possible role in that environment. As defined below metagenomic findings have resulted in restructuring of our understanding of the ecological balance in most of the environments.
7.3.1.1 Community Structure
Genomic information of the ‘unculturable’ bacteria can help in understanding their physiology and also their role in ecosystems. Metagenomic investigation has revealed that bacteriorhodopsins that function as light- driven proton pump for energy generation in halophilic archaea are also present in eubacteria. Presence of rhodopsin in marine proteobacteria suggested the possibility of a new phototrophic pathway that may influence the flux of carbon and energy in the ocean’s photozone worldwide. By utilizing shotgun sequencing approach complete genomes of Leptospirillum group II and Ferroplasma type II were assembled from a natural acidophilic biofilm. Pathways of carbon and nitrogen fixation and energy generation for every organism of the simple biofilm community were established. The almost complete assembly of the genome of an uncultured bacterium, Kuenenia stuttgartiensis, has revealed unique metabolic adaptations associated with anaerobic ammonium oxidation. PCR-DGGE (Denaturing gradient gel electrophoresis) is a sophisticated method that enables us to visualize a complete microbial community as a simple banding pattern and also allows to easily monitor community dynamics within the environment. In addition to usual 16S rRNA, various functional genes have also been targeted to investigate the diversity and community structure in this approach. Sargasso Sea genome project carried out by Venter et al. (2004) has resulted in the identification of genes for transport of phosphonates and utilization of polyphosphates and pyrophosphates. The finding indicates the microbial community’s capability to survive in the extremely phosphate-limited environment of Sargasoo Sea.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


