Metabolic and Inflammatory Responses to Trauma and Infection
Naji N. Abumrad
Igal Breitman
Julia Wattacheril
William J. Hubbard
Irshad H. Chaudry
Introduction
Surgery has its roots in providing care for those patients coping with injury or infection. In the last decade, an enormous amount of data has been published, which describes the wide spectrum of illnesses that can result following trauma or infection—from a minor, local reaction to surgery, to a systemic stress response, to sepsis, to systemic inflammatory response syndrome (SIRS), and, finally, to multi-organ failure (MOF). This information has provided the basis for many new concepts and techniques, which are now used daily in modern surgery. Having a thorough understanding of the mechanisms leading to illness following trauma and infection is crucial for any practicing surgeon. This understanding is the very hallmark of transferring knowledge gained in research to innovative surgical care at the bedside.
Overview
Following extensive tissue damage or systemic insult, such as infection, hypoperfusion, hypothermia, acid–base disturbance, pain or severe emotional stress, various physiologic and biochemical local and systemic alternations can be present and are referred to as “the stress response”. The systemic alternations are mediated by a complex signaling system, including afferent and efferent nervous signals, immunological and hormonal adaptations, and a systemic washout of locally produced substances like cytokines and other mediators. The first reference to the stress response resulted from keen observations by Sir David Cuthbertson in the 1930s who described a biphasic immune, inflammatory, and metabolic response to injury. This was further modified by Francis Moore in the 1970s. The first short (<24 hours) hypometabolic phase (termed “Ebb” by Cuthbertson) represents a coordinated response directed toward immediate survival. It starts with the activation of local coagulation and innate immune system factors. While evidence of a systemic response may be minimal in subjects with mild injury, in an insult of sufficient magnitude, the local activation is followed by systemic inflammatory and endocrine responses. These can present as surges in plasma catecholamine, cortisol and aldosterone levels inflicting tachycardia, tachypnea, vasoconstriction, lower cardiac output, lower oxygen consumption, lower basal metabolic rate, sodium and water retention, translocation of blood from the peripheral to the central vital organs, and acute-phase protein (APP) production. If the organs survive, there is transition from the “ebb” phase to the “Flow” phase. The flow phase of the stress response is characterized by explosive metabolic activity, increasing immune activity, enhanced enzymatic activity, and tissue repair. This response is mediated by a massive neuroendocrine flux involving the production and secretion of catecholamines, antidiuretic hormone (ADH), cortisol, insulin, glucagon, and growth hormone (GH). The increased adrenergic stimulation causes an increase in the ratio of glucagon to insulin and, combined with the increased cortisol and cytokines, induces the state of enhanced proteolysis and lipolysis.
The supply of amino acids comes from catabolism of mostly skeletal muscle and visceral organs. Some of these amino acids are taken up by the liver as substrates for gluconeogenesis and protein synthesis. Others are reserved for enzyme synthesis and collagen deposition at the site of injury. The energy needs of most other tissues are met by the availability of free fatty acids (FFA) and ketone bodies. These are made available via enhanced lipolysis with released glycerol acting as a glucose precursor. The hepatic glucose production supplies the glucose obligatory tissues.
Clearly, this process of catabolism requires an enhancement of blood flow to the muscle, the liver, and the areas of injury. Individuals present with tachycardia and tachypnea, peripheral edema, fever, hyperglycemia, leukocytosis, increased O2 consumption, increased CO2 production, increased minute ventilation, elevated resting energy expenditure and negative nitrogen balance. Consequently, the liver provides substrates through gluconeogenesis and synthesis of ketone bodies, detoxifies nitrogenous waste via the synthesis of urea and elaborates a series of APPs that bind metabolic by-products or limit the activity of proteolytic enzymes secreted by activated leukocytes. Renal blood flow and glomerular filtration increase and facilitate excretion of the nitrogenous by-products. Cytokines released from macrophages and adipokines released from adipose tissue result in disruption of capillary tight junctions, leading to vascular leak allowing fluid and substrates to flow toward the avascular area of injury, as well as to the interstitium in other body parts.
Manifestations of this hypermetabolic phase can be seen clinically in every postoperative patient. Patients retain fluid and sodium via concentrated urine, and redistribute blood flow to the vital organs, as well as compensate for the intravascular depletion secondary to capillary leak and possible external losses. If allowed to go unchecked, this catabolic response would deplete endogenous resources and become maladaptive. Systemic inflammatory response, severe metabolic depletion, and possible secondary infection can all cause damage to vital organs that were not initially compromised by the injury. Adult respiratory distress syndrome (ARDS), renal insufficiency, hepatic dysfunction, loss of gut epithelial barrier function, immunoparalysis, and sepsis may develop and the multi-organ dysfunction can be fatal. Fortunately, with appropriate support measures, the stress response nearly always resolves itself without complications.
The intensity and duration of the flow phase roughly correlate to the extent and type of injury. The catabolic process usually peaks at about 48 to 72 hours post-injury. If the insult is resolved, it can lead to an anabolic state, dominated by insulin, GH, and insulin-like growth factor I (IGF-I) within 5–10 days of injury. The change is associated with a flux of protein, fluid, and electrolytes returning to depleted intracellular space, particularly the muscle. Interstitial edema fluid is reabsorbed and the excess fluid is eliminated with a brisk diuresis. As the cellular space re-expands, the need for electrochemical equilibrium mandates the movement of ions (K+, Mg2+ and PO42-) from the blood into the cells. Serum levels of these ions decrease and require repletion. Anorexia and fatigue gradually resolve, and heart rate, respirations, and plasma glucose normalize. Nitrogen balance becomes positive and homeostasis is restored.
Inflammatory Response
The inflammatory response to injury involves interplay between several hormones (catecholamines and cortisol) and a large number of mediators (cytokines and chemokines). The immune and inflammatory responses to injury are predictable and well-orchestrated, and adaptive series of events evolve leading to maximize healing potential. A normal, balanced, and well-controlled inflammatory response in previously healthy patients almost always results in an uneventful recovery.
Innate Immune System
The immune response can be divided into an early innate and a later adaptive responses. The innate immune system is the first line of defense and its principal components are the epithelial barriers, immune cells (phagocytes such as neutrophils, macrophages and dendritic cells, and natural killer [NK] cells). Tissue damage, or microorganisms invading one or more of the epithelial barriers, is immediately recognized by the multiple components of innate immunity. The mechanisms used by the innate immune system to recognize nonself entities have been elucidated only recently. The innate immune response derives from preexisting recognition of pathogen-associated molecular patterns (PAMPs) or microorganism-associated molecular patterns (MAMPs). The best known examples of PAMPs are lipopolysaccharides (LPS) in gram-negative bacteria, lipoteichoic acids in gram-positive bacteria, mannose-rich oligosaccharides in microbial glycoproteins, mannans, unmethylated CpG sequences in bacteria, double-stranded RNA in replicating viruses, glucans, and N-formylmethionine (bacterial eukaryotic protein).
The receptors that have evolved to recognize these PAMPs are called pattern-recognition receptors, and these can functionally be divided into endocytic receptors, which mediate internalization and phagocytosis of microbes, and signaling receptors, which activate cellular signaling pathways that induce the expression of a variety of immune-response genes. The most important receptors that mediate endocytosis are the mannose receptors of the calcium-dependent lectin family, which recognize terminal mannose and fucose residues of glucoproteins and glycolipids that are characteristic of microorganisms, as well as the scavenger receptors that bind to bacterial cell walls. Among these signaling receptors, the two main groups of receptors are the Toll-like receptors (TLRs) and the G-protein-coupled receptors, of which the TLRs are the most prominent in the induction of immune and inflammatory responses.
Toll-like Receptors
The Toll signaling pathway was initially described in Drosophila in 1985, with the human homologue identified in 1997. This family of type I transmembrane receptors is characterized by an extracellular domain with leucine-rich repeats and a cytoplasmic domain. At least 11 human TLRs have been identified, and each is known to detect a specific PAMP and has a specific intracellular signaling pathway. TLR-1, 2, 4, 5, and 6 mainly recognize bacterial products, of which TLR2 has been implicated in the signaling process of gram-positive bacteria. TLR4 is the main receptor mediating the proinflammatory cytokines’ response to LPS. TLR-3, TLR-7, and TLR-8 are specific for viral detection and TLR-9 seems to be involved in both microbial and viral recognition.
TLRs seem to play a bridging role between the innate and the adaptive immune systems. They are expressed on dendritic cells and T-lymphocytes, as well as on a variety of parenchymal cells (e.g., adrenals, liver, and spleen). The adrenal-expressed TLRs influence the systemic inflammatory response by their effect on cortisol secretion. Upon sensing danger, the TLRs are activated on immune, competent, and endothelial cells ultimately resulting in the translocation of nuclear factor (NF)-κB. NF-κB then migrates to the nucleus and mediates gene transcription and the production of inflammatory mediators, such as chemokines, adhesion molecules, growth factors, and pro-inflammatory cytokines, especially tumor necrosis factor-α (TNF-α) and interleukin-1 (IL-1). The IL-1 and TNF-α receptors, after binding to their ligand, can further activate the same signaling pathways amplifying the immune response (Figure 1). TLRs are also involved in the recognition of endogenous ligands, which are released from damaged or dying cells, or come from a depredated extracellular matrix. These molecules include lipids, carbohydrates, proteins, and nucleic acids.
Extensive research has been conducted on whether genetic variations can be used to identify patients at high risk of developing sepsis and organ dysfunction during severe infection. Increasing evidence suggests that a genetic polymorphism in TLRs may influence a patient’s outcome in sepsis. For example, a single nucleotide polymorphism of TLR1 (TLR1-7202A/G) has been associated with higher organ dysfunction, increased gram-positive infections, and death by sepsis. Patients with a TLR4 gene mutation, especially those involving TLR4, Asp299Gly allele have a higher incidence of gram-negative infections; this polymorphism is also attributed to the severity of SIRS. Although septic patients with TLR-4 polymorphism have been shown to have reduced levels of circulating inflammatory cytokines and an increased risk of bacterial infection, the associations of mortality with polymorphism in TLRs during sepsis are still controversial. New research suggests that manipulation of TLR signaling pathways offers significant therapeutic potential, particularly in the treatment of organ injury accompanying sepsis, but this concept requires further exploration.
G-Protein-Coupled Receptors
These receptors initiate intracellular responses through the associated guanosine triphosphate (GTP)-binding G protein. These receptors are activated by chemokines, proteolytic products of complement proteins (e.g., C5a), and lipid mediators of inflammation (platelet-activating factor, prostaglandin E, and leukotriene B4).
Complement
The complement system consists of more than 30 proteins, including serum, serosal, and cell membrane proteins. Being part of the innate immune system, the complement system does not require prior immunization for activation; it is rapidly activated in a nonspecific manner in one of three main pathways: classic, alternative, and mannan-binding lectin pathways. In the classical pathway, it is activated by an IgM or IgG antibody–antigen complex. The alternative pathway does not rely on an antibody–antigen complex; it is activated directly by bacterial cell wall components. The mannan-binding lectin pathway is homologous to the classical pathway, except that the cascade is initiated by a mannan-binding lectin protein, produced by the liver that can activate complement cleavage when binding to a pathogen surface. Activation of the complement cascade results in the formation of products that act to lyse microbes, activate platelets, stimulate histamine release, recruit neutrophils by chemotactic action, and facilitate both phagocytosis and bacterial killing through opsonization of bacteria and stimulation of neutrophil degranulation.
Complement activation pathways are regulated by a large number of regulatory complement-control proteins, preventing over-activation of the whole system; systemic overwhelming activation of the system can result in changes in hemodynamic parameters, leading to shock. Persistent elevation of the complement-derived chemotaxins C3a, C4a, and C5a have been correlated with increased remote organ damage and
mortality following sepsis. Neutralization of C5a, using a monoclonal antibody, resulted in improved survival and decreased organ damage in animal models.
mortality following sepsis. Neutralization of C5a, using a monoclonal antibody, resulted in improved survival and decreased organ damage in animal models.
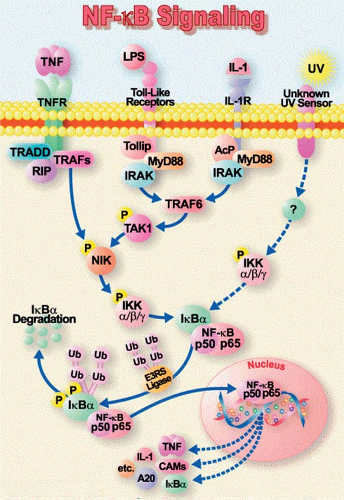 Fig. 1. The proinflammatory signal transduction pathway. ACP, accessory membrane-spanning protein; IKKα, inhibitory protein IκB kinase α; IKKβ, IκB kinase β IL-1, interleukin-1; IL-1R, IL-1 receptor; IRAK, IL-1R-activated kinase; NIK, NF-κB-inducing kinase; RIP, receptor-interacting protein; TNFR, tumor necrosis factor receptor; TRADD, TNFR and associated death-domain protein; TRAF2, TNFR-associated factor 2. (Modified from Baeuerle PA. Pro-inflammatory signaling last pieces in the NF-kappaB puzzle? Curr Biol 1998;8:R19, with permission.) |
Alarmins
Activation of the immune system is triggered by injury or trauma without evidence of a bacterial focus. This is mediated by alarmins or PAMPs. The alarmins are released either after a nonprogrammed cell death or by cells of the immune system. Within this family of endogenous triggers are high mobility group box 1 (HMGB1), heat shock proteins (HSPs), defensins, cathelicidin, eosinophil-derived neurotoxin (EDN), and others. These structurally diverse proteins serve as endogenous mediators of innate immunity as chemoattractants and activators of antigen-presenting cells.
HMGB1 was originally identified as a chromatin-binding protein that exists ubiquitously in the nucleus of all eukaryotic cells. HMGB1 plays a critical role in stabilizing nucleosome formation and in regulating transcription; it also plays an important role in signaling following tissue damage. When present in an extracellular location, HMGB1 can activate the innate immune system and promote inflammation. It is passively released by necrotic, but not apoptotic cells as well as actively secreted by immune cells, macrophages, and NK cells upon activation with TNF. HMBG1 acts as a chemokine and is a chemoattractant for macrophages, neutrophils, and dendritic cells and causes the secretion of several proinflammatory cytokines (e.g., TNF, IL-1a, IL-1b, IL-1RA, IL-6; IL-8, MIP-1a, and MIP2b) (Figure 2).
The role of HMGB1 in multi-organ damage in severe sepsis was demonstrated in an animal model. Inhibition of HMGB1 by specific antibodies protected mice from mortality in both LPS-induced and cecal ligation and puncture-induced sepsis. Furthermore, administration of recombinant HMGB1 protein recapitulated severe sepsis by inducing lethal organ dysfunction. Several techniques have been developed to inhibit the biological activity of HMGB1 in sepsis. A protein fragment A-box, which contains the DNA-binding domain of HMGB1, competes with intact HMGB1 for binding to its cell surface receptor, and exhibited a therapeutic effect in sepsis models even when administered after the onset of the diseases. Ethyl pyruvate, a stable and nontoxic derivative of pyruvic acid, has been shown to suppress HMGB1 release from macrophages in vitro, reduce serum HMGB1 levels, and improve survival in sepsis models in mice.
Adaptive Immune System
Adaptive immunity constitutes the second, but more specific and efficient response to invaders. It is subdivided into cellular and humoral immunity.
Cellular Immunity
Surgical insult leads to the activation of local host responses necessary for protection against invading microorganisms and for the initiation of tissue repair. The sequence of events begins immediately after injury, with the activation of the coagulation cascade and the initiation of the inflammatory phase. Local mediators of inflammation, such as cytokines, histamine, kinins, and arachidonic acid metabolites, cause increased capillary permeability, allowing immune cell infiltration (primarily neutrophils, followed by monocyte/macrophages
infiltration). Immune cell migration is a complex process involving attachment to the endothelial cells and extravasation regulated by many substances, the most important of which are the chemokines and adhesion molecules. Most of these mediators act in a paracrine fashion and they are short-lived because of rapid metabolism. Therefore, serum measurements of these mediators may not reflect their activity in local tissues.
infiltration). Immune cell migration is a complex process involving attachment to the endothelial cells and extravasation regulated by many substances, the most important of which are the chemokines and adhesion molecules. Most of these mediators act in a paracrine fashion and they are short-lived because of rapid metabolism. Therefore, serum measurements of these mediators may not reflect their activity in local tissues.
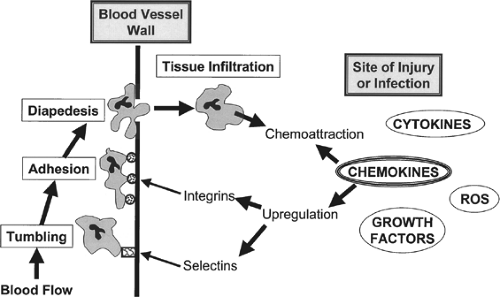 Fig. 2. Schematic of the chemokines-mediated process of polymorphonuclear leukocyte (PMN) recruitment and infiltration. Chemokines emanating from a source of injury and/or infection mediate the positively regulated expression of adhesion molecules. In this example, selectins and integrins cause tumbling and adherence, respectively, of PMNs to the endothelial lumen wall. The adherent PMN then moves through the wall by diapedesis and migrates along the chemokines gradient, eventually infiltrating in and around the focus of injury. ROS, reactive oxygen species. |
TLR activation causes secretion of cytokines (TNF-α and IL-1) and chemokines, especially by local macrophages. Chemokines are produced and secreted to the extracellular matrix by activated leukocytes and by various skin cells (epithelial cells, fibroblasts, and endothelial cells), and mediate cell motility. The local microcirculatory inflammatory response is reflected by a pronounced leukocyte accumulation and adherence to the endothelial lining of postcapillary and collecting venules. This response is associated with an increase in microvascular permeability, indicating the disruption of endothelial integrity.
Cytokines act on endothelial cells and induce the adhesion molecules. Leukocytes express carbohydrate ligands to bind to E and P endothelial selectins (a family of three single-chain transmembrane glucoproteins, named L, E, and P selectins), a process called “tethering.” These are low-affinity interactions and the leukocytes begin to roll along to the endothelial surface due to the force of the flowing blood. Chemokine signaling on rolling leukocytes results in the modification of the structure of a family of transmembrane proteins called “integrins,” allowing for firm adherence of leukocytes to the endothelial surface. Chemokines can then stimulate the extravasation and migration of the cells to the wound space.
Finally, at the time of injury, the production of pro-inflammatory cytokines and the expression of E-selectin, chemokines, and integrin ligands on endothelial cells mediate the selective recruitment of cutaneous lymphocyte antigen (CLA)-positive T-cells into the wound. There, they recognize the antigen for which their receptor is specific and become activated. The local macrophages act as antigen-presenting cells and also express the costimulatory molecules that are essential for T-cell activation. After antigen binding, T-cells differentiate preferentially into Th1 subsets, and secrete interferon-gamma (IFN-γ), the major macrophage-activating cytokine. The activated macrophages remove debris from dead cells to facilitate repair after the infection is controlled. The clearance of the debris and the infectious organisms promotes resolution of the inflammatory phase and ensuing repair responses, which include formation of granulation, reepithelialization, and neovascularization. The immune response then produces the cardinal signs of swelling, pain, erythema, and fever.
In the normal host response, these processes are mostly limited to the site of trauma; however, every substantial traumatic injury also leads to a degree of systemic inflammation. Depending on the magnitude of tissue damage, the local inflammatory process will cause washout of pro-inflammatory mediators into the systemic circulation and inflicts a systemic inflammatory process. The systemic leak of cytokines leads to further activation of immune cells, mostly polymorphonuclear leukocytes (PMN) priming, more cytokine secretion, activation of complement and the coagulation cascade, and secretion of APPs and neuroendocrine mediators. This systemic inflammation is followed by a compensatory anti-inflammatory response, creating a balance, which will have significant impact on the clinical outcome. Hence, the “right tuning” of systemic inflammation is crucial for restoration of homeostasis. Severe inflammation may lead to tissue destruction in organs not originally affected by the initial trauma by a process commonly referred to as the multiple-organ dysfunction syndrome (MODS). A lesser inflammatory response (or too much anti-inflammatory regulation) will induce a state of immunosuppression during the vulnerable time of recovery, which can result in deleterious sepsis for the host.
Cytokines
Cytokines are small proteins, secreted by systemic immune cells, macrophages, monocytes, or lymphocytes (mostly T-cells) and by diverse cell types at the site of injury. Cytokines are crucial mediators in cell immunity and inflammatory response. In healthy humans, they are produced at low constitutive levels, reaching just picograms per milliliter in plasma, and function in an endo-, para-, or autocrine manner. Cytokine receptors are expressed on the surface of the majority of human cells, and some soluble cytokine receptors are detectable in plasma at low levels. Cytokines activate intracellular signaling pathways that regulate gene transcription. Examples include NF-κB, activating protein 1 (AP-1), signal transduction- and transcription-activating factor 3 (STAT-3), and members of the CCAAT/enhancer binding protein (C/EBP) family of transcription factors, in particular C/EBP-β and δ.
The NF-κB family of transcription factors is most often studied because of its central role in the inflammatory process. Cytokines influence immune cell activity, differentiation, proliferation, and survival. These mediators also regulate the production and activity of other cytokines in a watershed manner. There is a significant overlap in bioactivity among different cytokines.
Cytokines are not antigen-specific and their effect can be stimulatory or inhibitory.
TNF, IL-1b, IL-6, IL-8, IL-12, and IFN-γ are the dominant stimulatory (or pro-inflammatory) cytokines and IL-4, IL-10, and IL-13 are considered inhibitory (or anti-inflammatory). Those compounds acting in between cells of the immune system are called interleukins, and those inducing chemotaxis of leukocytes are referred to as chemokines. Including about 50 chemokines and 30 interleukins, the number of characterized cytokines is now well in excess of 100. The number of cytokines recognized continues to grow, and a list of cytokines and their function(s), origin, target cells, and properties is provided in the Cytokine Online Pathfinder Encyclopedia (COPE) web site, created by Dr. Horst Ibelgaufts (www.copewithcytokines.de/cope.cgi).
TNF, IL-1b, IL-6, IL-8, IL-12, and IFN-γ are the dominant stimulatory (or pro-inflammatory) cytokines and IL-4, IL-10, and IL-13 are considered inhibitory (or anti-inflammatory). Those compounds acting in between cells of the immune system are called interleukins, and those inducing chemotaxis of leukocytes are referred to as chemokines. Including about 50 chemokines and 30 interleukins, the number of characterized cytokines is now well in excess of 100. The number of cytokines recognized continues to grow, and a list of cytokines and their function(s), origin, target cells, and properties is provided in the Cytokine Online Pathfinder Encyclopedia (COPE) web site, created by Dr. Horst Ibelgaufts (www.copewithcytokines.de/cope.cgi).
During acute localized inflammation, connective tissue, endothelial cells, and local immune cells are first to secrete pro-inflammatory cytokines, mostly IL-1 and TNF. Cytokines may leak to the circulation and exceed the levels of soluble receptors, which results in systemic inflammation and possible development of SIRS and MODS. The monocyte/macrophage also produces the only natural and well-characterized competitive cytokine antagonist, IL-1 receptor antagonist (IL-1ra), as well as liberates soluble forms of TNF and IL-1 receptors (IL-1RI) that are able to bind and neutralize TNF and IL-1, respectively. The t½ of circulating unbound cytokines can vary from <5 minutes to a few hours.
Interleukins
One of the best-described pro-inflammatory cytokines, TNF (previously known as cachectin) is mainly produced by macrophages and monocytes, and by T-cells, endothelial cells, fibroblasts, and adipose tissues. TNF is among the early cytokines secreted after trauma with a t½ < 20 minutes. TNF acts through its receptors TNFR1 and TNFR2.
TNF, through TNF-R1, activates the caspase cascade and induces cell apoptosis, as well as induction of transcription factors (e.g., NF-κB) and activation of the mitogen-activated protein kinase (MAPK) pathways both involved in cell proliferation, transcription of inflammatory genes, and anti-apoptosis. Binding of TNF to TNFR2 leads to activation and proliferation of immune cells. TNF induces secretion of a variety of pro- and anti-inflammatory cytokines (e.g., IL-6, IL-8, IFN-γ, and IL-10), increases synthesis of nitric oxide, activates the arachidonic acid pathway and induces activation of cyclooxygenase and lipoxygenase enzymes. This leads to the production of thromboxane A2 and prostaglandins E2, which have multiple physiological effects, including increased permeability of endothelial cells. It also induces the production of adhesion molecules, such as selectins, platelet-activating factors, and intracellular adhesion molecules (ICAM). In addition, TNF increases the pro-coagulated activity of endothelial cells.
The local effects of TNF can be physiologic, but the systemic effects often lead to adverse outcomes. TNF has been identified as a principal mediator in septic shock. In the central nervous system (CNS), TNF stimulates the release of corticotropin-releasing hormone (CRH), induces fever, and reduces appetite. In the liver, it stimulates production and secretion of APPs, and also causes insulin resistance. Inhibition of TNF by either anti-TNF antibodies or soluble receptors for TNF has become a strategy in the treatment of patients with chronic inflammatory diseases, but this strategy does not work in septic patients.
IL-1 was first described as endogenous pyrogen over a half century ago, because it caused fever when injected into rabbits. After being secreted by monocytes, macrophages, or endothelial cells, it has a t½ of only 6 minutes. The two forms, IL-1α and IL-1β, are regulated by different antigens, but both bind to the same IL-1RI. Binding to this receptor activates a signaling cascade that is shared also with IL-18 and TLRs. The IL-1 is a potent pyrogen, which influences the hypothalamus to reset the temperature of the body and induces fever. It is associated with local hyperalgesia. IL-1 has similar effects to TNF on the immune system following trauma. In fact, TNF and IL-1 are often described as synergistically acting mediators.
Similar to other cytokines, IL-6 is produced by a variety of cell types. It is detectable within an hour of trauma, and peaks at 4–48 hours following surgery. The secretion of IL-6 is induced by TNF and IL-1. IL-6 induces a proliferation and differentiation of B- and T-lymphocytes, activates NK cells and neutrophils, and inhibits its apoptosis. IL-6 regulates the hepatic synthesis of APP, such as C-reactive protein (CRP), fibrinogen, complement factors, α-2 macroglobulin, α1-antitrypsin, and others. IL-6 also induces the release of soluble TNF-R and IL-I receptor antagonist, and therefore plays a dual role in the inflammatory response by acting as both a pro-inflammatory and an anti-inflammatory mediator. IL-6 has a longer t½ than TNF or IL-1, which makes it easier to monitor, and seem to correlate with the magnitude of trauma. For example, despite similar procedure times, there is a greater degree of IL-6 elevation after abdominal aortic and colorectal surgery than after hip replacement; there are lower IL-6 levels after laparoscopic than after open procedures, including cholecystectomy and small-bowel and colonic resections. It has been shown in murine models that IL-6 is an important mediator of inflammation, and blocking IL-6 increases survival. Furthermore, IL-6 is regarded as a prognostic marker of trauma patients with SIRS, sepsis, or MODS and as such has been used in the intensive care unit (ICU) setting as an indicator for the severity of the inflammatory responses that is relatively independent of bacterial infections.
Chemokines
Overall, 18 chemokine receptors and 43 chemokines have been described, demonstrating a sharing of receptors. Chemokines acts as attractants to almost all blood cell types of the innate and adaptive immune response. In lower doses, chemokines act mostly as chemoattractants, while in increased concentrations they can lead to cell activation, including cytotoxicity and even respiratory burst. Their receptors have also been detected in endothelial cells, keratinocytes, and fibroblasts, suggesting that some chemokines also contribute to the regulation of epithelialization, angiogenesis, and tissue remodeling. The chemokine receptors belong to the family of G-protein-coupled receptors, and binding to these receptors leads to effects, including both chemotaxis and activation.
IL-8 is a typical chemotactic cytokine and its secretion is induced by IL-1, TNF-α, C5a, microbes and their products, hypoxia, hyperoxia, and reperfusion. Interferons attenuate the expression of IL-8. It can be produced in an early state of inflammation following trauma and can persist over a long period of time, even weeks. It has the ability to act as potent angiogenic factor, as a potent chemoattractant, and as an activator of immune cells. IL-8 signaling also induces the shedding of L-selectin from the neutrophil cell surface, and together with TNF-α and IL-6 is responsible for the regulation of adhesion molecules on endothelial cells. It is not the concentration of IL-8, itself, but the development of a concentration gradient that directs the cellular recruitment to the site of inflammation. There is also evidence that IL-8 can protect neutrophils against apoptosis, which could be one reason for prolongation of the inflammatory response at the site of injury or infection. It has also been shown that IL-8 plays an important role in the development of the ARDS.
Recently, a group of so-called silent chemokine receptors has gained more attention. These receptors can bind chemokines,
but do not evoke chemokine-related cell responses, suggesting a role as decoy or scavenger receptors. One member of this family is the decoy receptor D6. D6 binds most inflammatory chemokines, except IL-8, and is now known to be important in limiting the inflammatory response in different animal models, allowing degradation of chemokines. Another important member is the Duffy Antigen Receptor for Chemokines (DARC). DARC, first described as a blood group antigen, is also expressed by red blood cells and endothelial cells. It binds angiogenic chemokines, including IL-8. While D6 eliminates the cellular response to chemokines, the DARC receptor seems to act more to differentiate this response, and chemokines retain their biological activity after binding to this receptor. DARC on red blood cells seems to capture chemokines and is, therefore, supposed to prevent leukocyte activation in the systemic circulation. On the other hand, DARC on endothelial cells is required for leukocyte recruitment.
but do not evoke chemokine-related cell responses, suggesting a role as decoy or scavenger receptors. One member of this family is the decoy receptor D6. D6 binds most inflammatory chemokines, except IL-8, and is now known to be important in limiting the inflammatory response in different animal models, allowing degradation of chemokines. Another important member is the Duffy Antigen Receptor for Chemokines (DARC). DARC, first described as a blood group antigen, is also expressed by red blood cells and endothelial cells. It binds angiogenic chemokines, including IL-8. While D6 eliminates the cellular response to chemokines, the DARC receptor seems to act more to differentiate this response, and chemokines retain their biological activity after binding to this receptor. DARC on red blood cells seems to capture chemokines and is, therefore, supposed to prevent leukocyte activation in the systemic circulation. On the other hand, DARC on endothelial cells is required for leukocyte recruitment.
Cytokines Post-elective Surgery
Elective surgery followed by an uneventful clinical course may induce only minor systemic inflammatory changes. As one could expect, the acute-phase response, post-elective surgery, is proportional to the surgery-related tissue trauma or to the severity of the procedures. Virtually, all mediators of inflammation (cellular, cytokines, and APPs) peak post-injury at about day 1 to 2 and then return to baseline levels by post-injury days 6 to 7. Persistent postoperative pain, stress, or a second insult will change that pattern.
The Neuro-Immune Axis
The systemic and even local inflammatory responses posttrauma are regulated by the nervous system. Considerable attention has been given to the effectiveness of parasympathetic nerve stimulation in suppressing the magnitude of the proinflammatory response, leading to coining of the term “inflammatory reflex.” Like other reflex arcs, the inflammatory reflex is comprised of a sensory afferent arm and an efferent motor arm.
Afferent/Sensory Input to the Brain
During stress, afferent signals from the injury site can reach the CNS through two main routes: the neural route, mostly by afferent vagal fibers, and through blood-borne inflammatory mediators.
Neural Route
The neural afferents present a rapid means to activate the CNS; the mechanism of their activation remains unclear. Various investigators demonstrated the effects of complement (C5a) fragments, PGE2, coagulation factors (Factor XII), kinins (bradykinin), and cytokines (TNF, IL-1, and IL-6). Increasing evidence has suggested that vagal pathways are utilized as the communication link between the peritoneal cavity and the CNS, especially during episodes of intraabdominal infection. It has been shown that many CNS effects induced by intraperitoneal administration of LPS or IL-1 (fever, increased elaboration of adrenocorticotrophic hormone [ACTH]) can be blocked or attenuated by subdiaphragmatic vagotomy. This sensory arm can be activated by the presence of IL-1 in peripheral tissues. Specific IL-1 binding sites have been revealed on glomus cells adjacent to the vagus nerve. IL-1 binding and an intact vagus nerve are both required for the development of fever following intraperitoneal administration of low quantities of IL-1.
Humoral Route
Cytokines are lipophobic molecules and do not have ready access to the CNS, since the blood brain barrier (BBB) excludes entry of such proteins. An exception is in regions where the BBB is not well formed, such as around the circumventricular organs (CVOs), the meninges and the choroid plexus. There may be active transport into specific regions of the brain of circulating cytokines by the vascular endothelium. Alternatively, cytokines may damage the integrity of the vascular endothelium that forms the BBB, enter the brain, and stimulate central neural circuits. Several factors have been implicated, most notably IL-1 and IL-6. Prostaglandins, mostly PGE2, locally produced in the hypothalamus in reaction to cytokines, play a crucial role in inducing pyrexic reaction, as known for many years from the ability of cyclooxygenase inhibitors to prevent fever.
Efferent Regulation
Following integration of afferent signals the CNS has two major effector/efferent arms that are used to regulate physiologic responses. The first is the activation of the hypothalamus-pituitary-adrenal (HPA) axis, and the second is the direct activation of the sympathetic system while suppressing the other parasympathetic “half” of the autonomic nervous system. The CNS regulates the “level of ongoing inflammation” through multiple pathways, both pro- and anti-inflammatory. The anti-inflammatory effect was studied more thoroughly. The inflammatory opposing response suppresses the immune system through at least two main routes: by increasing corticosteroid hormone levels (activation of the HPA axis) and by activation of the cholinergic anti-inflammatory pathway. Evidence exists that hormones and cytokines interact at several levels. For example, TNF-α, IL-1, and IL-6 stimulate the HPA axis resulting in the release of ACTH and glucocorticoids (Figure 3). IL-1 also has a direct enhancing effect on the adrenals. The glucocorticoids secreted down-regulate cytokine release from macrophages in a negative feedback mechanism. The negative feedback between glucocorticoids and cytokines is one of the main mechanisms protecting the organism from the possible damage from over inflammation.
Reduced triiodothyronine (T3) levels after treatment with TNF-α or IL-1 demonstrate another link between hormones and cytokines. Hormones can also influence each other, as catecholamines increase cellular uptake of T3. The “low T3 syndrome” in sepsis and following trauma may be due to a combination of cytokine and catecholamine effects. The complex interactions among different mediators may explain, at least in part, why treatment directed against individual mediators following trauma or sepsis has not been successful.
Immunosuppression Following Trauma
More and more evidence is emerging that the neurologic system plays a major role in the coordination of inflammatory and anti-inflammatory immune response. While minor surgery is suggested to stimulate components of the immune system, it is generally agreed that after the acute-phase response, major surgery, and to a higher extent, major trauma cause immunosuppression that may render the host anergic to opportunistic infections. The initial response to surgical trauma is characterized by activation of the specific and nonspecific immune system’s release of pro-inflammatory cytokines (TNF, IL-1β, IL-6, IL-18, and HMGB1 and more), neutrophil activation, microvascular adherence, as well as PMN and macrophage oxidative burst, but this rapidly gives way to a state of depressed immune function.
The production of immunoglobulins fall and many patients become anergic as assessed by delayed hypersensitivity skin testing. Defects in neutrophil chemotaxis, phagocytosis, and lysosomal enzyme content and respiratory burst have all been reported. This condition is referred to as a compensatory anti-inflammatory response syndrome; it is induced by multiple mediators and affects all subtypes of immunity. The counter anti-inflammatory mechanism is as complex and multi-factorial as the pro-inflammatory one. It includes cytokines
such as IL-10, TGF-β, TNF-binding protein, and hormones such as corticosteroids, adrenaline, and α-melanocyte stimulating hormone (α-MSH). These act in concert with local effectors, such as PGE2, HSPs, and APPs. These factors interact to inhibit macrophage activation and down regulate the synthesis of pro-inflammatory cytokines.
such as IL-10, TGF-β, TNF-binding protein, and hormones such as corticosteroids, adrenaline, and α-melanocyte stimulating hormone (α-MSH). These act in concert with local effectors, such as PGE2, HSPs, and APPs. These factors interact to inhibit macrophage activation and down regulate the synthesis of pro-inflammatory cytokines.
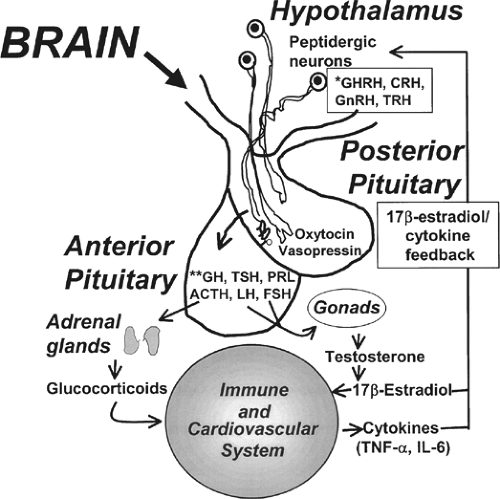 Fig. 3. Relationship between the hypothalamus–pituitary–gonad-adrenal (HPA) axis and the immune system in physiological responses to injury. The HPA is a neuroendocrine system that also has bidirectional communication with the immune system in homeostasis and in times of injury, giving the brain a major role in regulating endocrine and immune functions. The hormonal responses are apparent at three levels: the hypothalamus, the pituitary, and the adrenals. It can be seen that organs are coupled with one another (functioning as a biologic oscillators), with the coupling being mediated by neural, hormonal, and cytokine networks. Notably, cytokines and sex hormones are closely coupled in a counterregulatory fashion, which sheds light on the beneficial effects of sex hormones, especially β-estradiol, in responses to injury. |
The Cholinergic Anti-inflammatory Pathway
The activation of the cholinergic pathway leads to acetylcholine release in the reticuloendothelial system that includes the spleen, liver, lymphoid tissue, and GI tract. Acetylcholine binds to an α7 subunit of the nicotinic acetylcholine receptor, expressed on tissue macrophages, to inhibit the release of pro-inflammatory (TNF, IL-1β, IL-6, and IL-18), but not the anti-inflammatory cytokine IL-10. In macrophages, signaling through α7 attenuates TNF production through a mechanism dependent upon inhibition of NF-κB nuclear translocation and activation of Jak-STAT pathways. Direct electrical stimulation of the peripheral vagus nerve in vivo during lethal endotoxemia in rats inhibited TNF synthesis in liver, attenuated peak serum TNF amounts, and prevented the development of shock. Several reports have confirmed that the activation of this pathway, either by electrical stimulation of the vagus nerve or by administration of α7 selective drugs, is effective in ameliorating inflammation and improving survival in a number of experimental models, such as sepsis, hemorrhagic shock, pancreatitis, and postoperative ileus.
IL-6, As an Immunosuppressor
The massive and continuous IL-6 release accounts for the up-regulation of major anti-inflammatory mediators, such as glucocorticoids, PGE2, IL-10, and TGFβ. IL-6 stimulates the macrophage expression of anti-inflammatory mediators, such as IL-1RI antagonist and soluble TNF receptors. These bind to the pro-inflammatory cytokines TNFα and IL-1β and truncate the inflammatory response.
Cell-Mediated Immune Dysfunction
Cellular immuno-incompetence (also called “immune paralysis”) is induced by elevated PGE2, IL-10, and other anti-inflammatory mediators, mainly caused by the deactivation of monocytes. The central role of IL-10 and TGFβ in inducing monocyte “immune paralysis” is demonstrated by the up-regulation of HLA-DR expression on monocytes following the application of an IL-10 neutralizing antibody and the restoration of macrophage antigen presentation by using TGF-β neutralizing antibodies.
Lymphocyte Dysfunction
Major surgical interventions are associated with a significant decrease in total systemic lymphocyte counts, including both CD4+ and CD8+ cells. This lymphocyte depression correlates with the duration of the surgical procedure and the volume of blood loss, however, is not associated with the extent of the trauma, the age of the patient, or the type of intensive care intervention. These events are accompanied (within 24 h) with elevated IL-10 and increased frequency of apoptosis of CD4+ and CD8+ cells accompanied by marked down-regulation of anti-apoptotic factors such as Bcl-2. The impact of this immune dysfunction was underscored by the fact that the rate of apoptotic CD8+ cells significantly correlated with the manifestation of infectious complications during the postoperative course.
A considerable number of studies have shown that modulation of T-helper lymphocytes (Th cells) is also involved in the development of immune suppression following surgical trauma. The cells can be subdivided into two functionally distinct subsets: Th1 and Th2, according to individual functional parameters. Th1 cells may support an inflammatory response by producing IL-2, IL-12, and IFN-γ, while Th2 cells act as anti-inflammatory agents by secreting IL-4, IL-5, IL-6, IL-10, and IL-13. Major trauma is associated with a shift of the Th1/Th2 balance toward a Th2 response. Lymphocyte dysfunction may present as a complete lack of response to external stimuli, that is, anergy.
The Second Hit Phenomenon
The so-called two-hit model of inflammatory insult has become a commonly accepted paradigm. It takes place in many common scenarios in which the patient has to undergo a surgical procedure following initial trauma or suffers further insults due to a complication. The second hit may be
sterile- (operation after trauma) or pathogen-induced infection post-surgery.
sterile- (operation after trauma) or pathogen-induced infection post-surgery.
Although influenced by many factors, the inflammatory and metabolic response is relatively predictable. The immune reaction to further insults is not as consistent. Variations in the competence of innate and adaptive immune defenses become evident; there is an innate immune tolerance and diminished adaptive immune capacity of response to a new antigen. On the other hand, the recurrent immunological activation causes a persistent systemic pro-inflammatory activity that may lead to SIRS and MOF. The persistent inflammation could take place only in some aspects of immunity and not in others. An example of this is the, continuation of coagulation system activation, even while other pro-inflammatory activity is waning. Not infrequently, a prolonged stress state manifests diminishing amplitude, frequency, and efficiency of autonomic and neuroendocrine signaling. Disturbances in circadian rhythmicity of neuroendocrine hormone secretion are also observed during prolonged inflammatory illness. The attenuated hormone rhythmicity and signal amplitude may contribute to disordered metabolic and immune functions.
Systemic Inflammatory Response Syndrome and Multiple Organ Dysfunction Syndrome
Cytokine-mediated inflammation is usually short-lived and is resolved. In some cases, however, cytokine production can become excessive, and rather than resolving, inflammation persists or even spreads, causing damage in adjacent tissues. This hypermetabolic response, often called the SIRS, encompasses excessive whole body inflammation and is considered a major determinant in the development of multiple organ dysfunctions (MODs), often with a lethal result. The pathophysiology of SIRS and MODS is explored in Chapter 8.
Endocrine Response
Role of the Central Nervous System
The CNS response consists primarily of three parallel, coordinated effects: fever, HPA axis activation, and sickness behavior (such as anorexia or somnolence). Following integration of afferent signals, the hypothalamus has two major effector arms that are used to regulate physiological responses. The first is the activation of the HPA axis and the second is the direct activation of the sympathetic system, while suppressing the other parasympathetic “half” of the autonomic nervous system. At rest, the hypothalamus secretes, in a pulsing manner, CRH, thyrotrotropin-releasing hormone (TRH), gonadotropin-releasing hormone (GnRH), growth hormone-relapsing hormone (GHRH), and dopamine. During stress, the afferent signals from the injury site reach the hypothalamus through the neural route (impulses are transferred from the cephalad to the ventral-posterior nucleus of the thalamus) mostly by afferent vagal fibers or through blood-borne inflammatory mediators. Humoral mediators reach the hypothalamic-hypophysial portal capillaries in the median eminence through the anterior hypophyseal arteries. The cytokines can diffuse into the portal capillaries, areas that are free from the BBB.
Endogenous Opioids (Endorphins)
Many of the mediators released during inflammation of peripheral tissue are known to elicit pain by activation of specialized primary afferent neurons called “nociceptors” (defined as “neurons preferentially sensitive to a noxious stimulus or to a stimulus which would become noxious if prolonged”). Nociceptor stimuli propagate through the dorsal horn of the spinal cord to the supraspinal sites where a sensation of pain is eventually elicited. Various opioid peptides, such as β-endorphin, met-enkephalin, dynorphin, and endomorphins are produced and secreted by the hypophysis, hypothalamus, and, as demonstrated most recently, locally by leukocytes. Opioid peptides can bind to opioid receptors. The most studied opioid receptor groups are μ, κ, and δ. These receptors are part of the G-protein-coupled receptors, which are synthesized in dorsal root ganglia and are transported intra-axonally. The opioid receptors are represented in the brain, spinal cord, sensory peripheral nerve endings, and in the intestinal tract. Agonist binding elicits potent analgesia, a quality often used to treat pain, with induction of external opioids.
The balanced activation of sympathetic and parasympathetic pathways, as well as HPA axis, in response to injury is crucial in dynamic regulation of a host’s defense mechanisms. The endorphins are part of the counterregulatory system activated in a state of shock. The opioids enhance the parasympathetic tone, balancing the increased sympathetic drive. A meta-analysis review of the literature concerning the use of opioid antagonist (Naloxone) in clinical setup indicates that opiate antagonist treatment does improve mean arterial blood pressure in shock patients. The mechanism involved in mediating the salutary effects of opiate antagonists has not been completely elucidated.
Immune cells carry all three opioid receptors. Opioids have been shown to modulate a number of aspects of the immune response, including antibody responses in vitro and in vivo, phagocytic cell function, NK-cell activity, chemokine-induced chemotaxis, the development and function of T-cells in the thymus, and cytokine and cytokine receptor expression. Opiate-mediated immune effects have been postulated to result from either direct interaction with opioid receptors on cells of the immune system or indirectly through the activation of opioid receptors within the CNS, and the resulting modulation of HPA axis (cortisol) and the sympathetic nerve system activities. Although alternations in various aspects of immune function in patients exposed to opioid treatment were demonstrated in clinical practice (post-elective abdominal surgery, orthopedic surgery, and in healthy volunteers), there are no actual prospective clinical studies exploring the possible interaction between exposure to opiates and rates of infection.
Hormonal Changes During Acute and Chronic Surgical Illness
There is a biphasic neuroendocrine response to critical illness. The acute phase is characterized by an actively secreting pituitary; whereas, in prolonged critical illness, there is a hypothalamic suppression of the neuroendocrine axes.
Glucocorticosteroids
In a stress-free healthy human, cortisol is secreted from the zona fasciculata of the adrenal cortex, according to a diurnal pattern. Cortisol release is controlled by ACTH produced by the pituitary, in turn under the influence of the hypothalamic CRH. Cortisol itself exerts negative-feedback control on both hormones. Approximately 10% cortisol is found free in the plasma. Of the remainder, 20% is bound to albumin, and 70% is bound to cortisol-binding globulin. Only the free hormone, however, is biologically active. Glucocorticoids exert their effects by binding to and activating an intracellular receptor protein. The cortisol–glucocorticoid receptor complex moves to the nucleus where it binds as a homodimer to DNA sequences located in the promoter regions of target genes. In addition, the cortisol–glucocorticoid receptor complex may affect cellular function indirectly by binding to and modulating the transcriptional activity of other nuclear transcription factors, such as NF-κB.
Cortisol During Stress
Cortisol levels usually rise in the early phase of critical illness. The excited neurons in the hypothalamus release CRH and arginine vasopressin (AVP) from their terminals
into the capillaries of the hypothalamo-hypophysial portal system. CRH and AVP act on CRH-1 and vasopressin-1β receptors on the anterior pituitary to stimulate ACTH secretion. Plasma ACTH levels rise directly due to increased secretion and due to resistance to or inhibition of the negative-feedback mechanism exerted by cortisol. Several of the elevated cytokines have been shown to modulate cortisol production, either by directly affecting the hypothalamus/pituitary (IL-1α, IL-1β, IL-6, and TNF-α) or by direct stimulation of the adrenal cortex (IL-1α, IL-1β, and IL-6). Cytokines can also influence glucocorticoid receptor numbers and affinity. During severe illness, corticosteroid-binding globulin levels are decreased, resulting in proportionate increases in the free hormone. The diurnal variation in cortisol secretion is lost in response to any type of acute illness or trauma. An appropriate activation of the HPA axis and cortisol in response to critical illness is essential for survival. The adrenal gland does not store cortisol; therefore, increased secretion arises due to increased synthesis of cortisol from its principal precursor, cholesterol.
into the capillaries of the hypothalamo-hypophysial portal system. CRH and AVP act on CRH-1 and vasopressin-1β receptors on the anterior pituitary to stimulate ACTH secretion. Plasma ACTH levels rise directly due to increased secretion and due to resistance to or inhibition of the negative-feedback mechanism exerted by cortisol. Several of the elevated cytokines have been shown to modulate cortisol production, either by directly affecting the hypothalamus/pituitary (IL-1α, IL-1β, IL-6, and TNF-α) or by direct stimulation of the adrenal cortex (IL-1α, IL-1β, and IL-6). Cytokines can also influence glucocorticoid receptor numbers and affinity. During severe illness, corticosteroid-binding globulin levels are decreased, resulting in proportionate increases in the free hormone. The diurnal variation in cortisol secretion is lost in response to any type of acute illness or trauma. An appropriate activation of the HPA axis and cortisol in response to critical illness is essential for survival. The adrenal gland does not store cortisol; therefore, increased secretion arises due to increased synthesis of cortisol from its principal precursor, cholesterol.
Cortisol Influence on Post-trauma Physiology
The stress-induced hypercortisolism fosters the acute provision of energy. Glucocorticoids increase blood glucose concentrations by increasing the rate of hepatic gluconeogenesis and inhibiting adipose tissue glucose uptake. Hepatic gluconeogenesis is stimulated by increasing the activities of phosphoenolpyruvate carboxykinase and glucose-6-phosphatase as a result of binding of glucocorticoids to the glucocorticoid response elements of the genes for these enzymes. Glucocorticoids also stimulate free fatty acid release from adipose tissue and amino acid release from body proteins. Major roles of these processes are to supply energy and substrate to the cell, which are required for the response to stress and repair to injury.
The rise in glucocorticoids also protects against excessive inflammation. The rise in glucocorticoids during acute illness plays a crucial role in preventing hazardous over-stimulation of the immune system, including lymphocytes, NK cells, monocytes, macrophages, eosinophils, neutrophils, mast cells, and basophils. Glucocorticoids decrease the accumulation and function of most of these cells at inflammatory sites. Most of the suppressive effects of glucocorticoids on immune and inflammatory reactions appear to be a consequence of the modulation of production or activity of cytokines, chemokines, eicosanoids, complement activation, and other inflammatory mediators. Glucocorticoids control mediator production predominantly through inhibition of transcription factors, such as NF-κB. Glucocorticoids also produce anti-inflammatory effects by enhancing release of factors, such as IL-1RI antagonist, soluble TNF receptor, and IL-10. Glucocorticoids also block the transcription of messenger RNA for enzymes required for the synthesis of some mediators (cyclooxygenase-2 and iNOS).
A rise in glucocorticoid concentrations plays an important role in improving hemodynamic levels, by inducing fluid and sodium kidney retention. Glucocorticoids are also required for the needed increased sensitivity of the cardiovascular system to vasoconstrictors. The reactivity to angiotensin II, epinephrine (Epi), and norepinephrine (Norepi) contributes to the maintenance of cardiac contractility, vascular tone, and blood pressure. These effects are mediated partly by the increased transcription and expression of the receptors for these hormones. Glucocorticoids are required for the synthesis of Na+, K+-ATPase, and catecholamines. The effects of glucocorticoids on synthesis of catecholamines and catecholamine receptors are partially responsible for the positive inotropic effects of these hormones. Glucocorticoids also decrease the production of nitric oxide, a major vasorelaxant and modulator of vascular permeability.
During surgical procedures, such as laparotomy, serum corticotropin and cortisol rise rapidly, peaking in the immediate postoperative period. The magnitude of the postoperative increase in serum cortisol concentration is correlated with the extent of the surgery. From a normal secretion rate of 10 mg/day, cortisol production rate increases to 75 to 150 mg/day following major surgery and can reach to 250 to 300 mg/day in severe stress. Unless there is a repeated insult, such as sepsis, the glucocorticoid concentrations decline to baseline levels over the next 72 hours. This decline can often be noticed clinically as increased diuresis, improved glucose control, and, occasionally, increased pain. In critical illness, the kinetics of the response differ from those mentioned above: pain, fever, hypovolemia, hypotension, and tissue damage all result in a sustained increase in corticotropin and cortisol secretion and a loss of the normal diurnal variation in these hormones. During severe illness, serum cortisol concentrations tend to be higher than even in patients undergoing major surgery (∼30 μg/dL vs. 40–50 μg/dL).
Adrenal Insufficiency
Critical illness is associated with activation of the HPA axis; however, many factors can impair the integrity of the HPA axis, such as blunt normal response leading to either transient or, rarely, permanent adrenal insufficiency. This scenario can lead to a potentially lethal condition. Refractory hypotension is the most common aspect of acute adrenal insufficiency. Adrenal insufficiency should be suspected in any critically ill patient who has persistent hypotension and hemodynamic instability that persists despite adequate fluid resuscitation and/or requires vasopressor support. Other nonspecific signs can include multiple organ dysfunction, otherwise unexplained hypoglycemia, hyponatremia, hyperkalemia metabolic acidosis, eosinophilia, hyperdynamic circulation, and other pituitary deficiencies (gonadotropin, thyroid, and diabetes insipidus). Recently, much attention had been focused on the so-called relative or functional adrenal insufficiency of critical illness, a condition defined as subnormal adrenal corticosteroid production in the absence of any structural defects of the HPA axis. The explanation for the development of this condition is hypothetical exhaustion of the secretory adrenocortical reserve as a result of ongoing near-maximal stimulation. Other contributing factors may include the suppression of cortisol and ACTH production by circulating cytokines and other inflammatory mediators, as well as the development of target tissue resistance to glucocorticoids and/or adrenal cortex resistance to ACTH action. Currently, the clinical significance of this condition is not clear and was only demonstrated in a setup of septic shock. Although corticosteroid replacement therapy might also be beneficial to patients who have other critical illnesses in which there is evidence of relative hypoadrenalism, no high-quality data from large randomized studies is available.
As mentioned earlier, there is no clear or current threshold definition for physiological “normal” and low cortisol plasma concentration during critical illness. Since about 90% to 95% of plasma cortisol is bound to protein, the routine decrease in cortisol-binding protein and albumin following critical illness makes it difficult to calculate and interpret the meaning of total cortisol concentration. While the plasma proteins are low and there is a peripheral increased resistance to cortisol, as often happens in critical illness, the free cortisol levels are not a reliable reflection of either total cortisol secretion or action. Many thresholds, below which adrenal insufficiency is likely to be present, have been suggested, ranging widely from 10 to 34 μg/dL. Many textbooks and published articles state that the normal circulating cortisol
response to stress is a level >18 to 20 μg/dL. However, the choice of 18 to 20 μg/dL is based primarily on the response to exogenous high-dose ACTH stimulation and the response to insulin-induced hypoglycemia in nonstressed patients.
response to stress is a level >18 to 20 μg/dL. However, the choice of 18 to 20 μg/dL is based primarily on the response to exogenous high-dose ACTH stimulation and the response to insulin-induced hypoglycemia in nonstressed patients.
The high-dose (250 μg) ACTH stimulation test, instead of random cortisol levels, is traditionally used in ICU setup and is regarded as the “gold standard” for adrenal testing. One should be aware, however, that although this test can be informative as it relates to adrenal ability to react to excessive ACTH (cortical reserve), it does not reflect the integrity of the entire hypothalamic pituitary adrenal axis. As a result, it may not be able to properly diagnose secondary adrenal insufficiency. A low-dose (1 μg) cosyntropin stimulation test has been used to a small extent to diagnose secondary insufficiency in ICU setup. Other methods that have been suggested include calculated free cortisol index, measuring salivary cortisol, or measuring other ACTH-dependent adrenal steroids (DHEA, DHEAS). None have yet become the gold standard in the clinical setup. The Surviving Sepsis guidelines currently endorse the use of corticosteroids (200 mg of hydrocortisone/day in divided doses) in patients who have refractory septic shock. Although a reasonable recommendation, the authors would point out that this is essentially based on a single study from a single center that has not yet been confirmed adequately.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


