Mantle Cell Lymphoma
Definition
Mantle cell lymphoma (MCL) is a clinically aggressive B-cell lymphoma associated with the t(11;14)(q13;q32) and cyclin D1 overexpression in almost all cases (1). Originally, MCL was defined using histologic criteria as a neoplasm composed of small to medium-sized lymphoid cells with variably irregular nuclear contours and lacking centroblasts. Subsequent immunophenotypic analysis showed that MCL is of B-cell lineage, CD5+ and CD23–. Currently, the t(11;14) and cyclin D1 overexpression have assumed importance equal to traditional criteria, and their presence supports the diagnosis of MCL in cases with atypical histologic or immunophenotypic features (e.g., blastoid variant MCL, CD5– tumors).
Synonyms
Historical Introduction
Lennert first described MCL as diffuse germinocytoma at a meeting in London, England in 1973 (5). As part of the Kiel classification published in 1974, the name was changed to centrocytic lymphoma (6). At that time, the neoplasm was thought to arise from centrocytes (small cleaved cells) in the germinal center. About the same time, Berard and Dorfman described lymphocytic lymphoma, intermediate grade of differentiation (IDL) (2). Their concept was that IDL cells had nuclear irregularities intermediate between the entities defined in the Rappaport classification as well-differentiated lymphocytic lymphoma [currently chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL)] and poorly differentiated lymphocytic lymphoma [currently follicular lymphoma (FL) grade 1]. Subsequently, Weisenburger and colleagues (3,4) described intermediate lymphocytic lymphoma and mantle zone lymphoma, corresponding in large part to the diffuse and mantle zone patterns of MCL. When the Working Formulation was published in 1982, no specific category was designated for the lymphoma we currently designate as MCL (7). It was recognized in that study, however, that most cases of centrocytic lymphoma fit best within the diffuse small cleaved cell category. Small subsets of MCL were “squeezed” into other categories: typical cases were placed particularly within the small lymphocytic lymphoma and follicular small cleaved cell lymphoma categories. Blastoid variants, not recognized as MCL at that time, most likely were classified as lymphoblastic or diffuse large-cell lymphoma. Throughout the 1980s, Europeans used the Kiel classification and continued making the diagnosis of centrocytic lymphoma. In the United States, the Working Formulation was used, often with additional comments stating that the neoplasm also could be classified as IDL, intermediate lymphocytic lymphoma, or mantle zone lymphoma.
The t(11;14)(q13;q32) was first recognized as an uncommon cytogenetic abnormality in the 1970s and, with time, this translocation became associated with centrocytic lymphoma or IDL (8). In 1984, Tsujimoto and colleagues cloned the breakpoints of the t(11;14) and showed that the B-cell lymphoma leukemia-1 (bcl-1) locus and IgH were involved (9). In 1990, an abstract submitted to the United States and Canadian Academy of Pathology Annual Meeting from the National Institutes of Health (NIH) first reported bcl-1 rearrangements in IDL (10). This was followed by two papers in the journal Blood, the first by Williams and colleagues and the next by the NIH group reporting bcl-1 rearrangements in centrocytic lymphoma and IDL, respectively (11,12). At this point, it became clear (although it had been thought to be true) that centrocytic lymphoma and most IDL cases overlapped and were the same. Subsequently, ccnd-1 was identified as the gene on 11q13 involved in the t(11;14) (13). In 1992, a large group of hematopathologists issued a consensus statement on the criteria for this neoplasm and recommended use of the term mantle cell lymphoma (14). Mantle cell lymphoma is thought to arise from a CD5+ precursor within the inner half of the mantle zone of the lymphoid follicle, but the exact cell of origin in unknown (1,14).
Additional molecular studies have begun to unravel the molecular pathogenesis of MCL. These studies also have enabled pathologists to refine the criteria for MCL and have expanded the morphologic spectrum of this neoplasm. As a result, most B-cell lymphomas that carry the t(11;14), even if they have morphologic features atypical for MCL and most likely would have been classified as another type of lymphoma in the past, are currently thought to be best classified as MCL.
Epidemiology
In the Non-Hodgkin’s Lymphoma Classification Project, MCL represented 6% of all non-Hodgkin lymphomas (15). The relative frequency of MCL in other studies has ranged from 3% to 10%, and MCL may be relatively more common in Europe (1). The frequency of MCL in the Surveillance, Epidemiology, and End Results (SEER) database from 1992–2001 was 1.5% (16). We suspect that MCL was under-represented during this interval because consensus criteria were published in 1992, and cyclin D1 immunohistochemical staining was not available until a few years later. Affected patients are usually elderly, and MCL is much more common in men than women, by a ratio of 2.5 or 3:1. The incidence is higher in whites than African or Asian Americans (15,16).
Pathogenesis
The t(11;14)(q13;q32), which juxtaposes ccnd-1 with IgH on the derivative chromosome 14 and results in cyclin D1 overexpression, is a characteristic finding in MCL. The etiology for the genesis of the t(11;14) is uncertain, but sequence analysis suggests that this translocation arises as a mistake during immunoglobulin (Ig) gene rearrangement, during attempted V-D-J recombination (17). Some data also suggest that the ccnd-1 and IgH loci are in close spatial proximity in immature B cells, presumably thereby predisposing the cell to the occurrence of this translocation (18).
The t(11;14) is thought to be an early event in the pathogenesis of MCL that appears necessary, but not sufficient, for oncogenesis. This concept is supported by transgenic mice experiments. In one study, transgenic mice with cyclin D1 linked to Ig regulatory elements did not develop lymphoma (19). In a second similar study, when a second gene abnormality was also introduced, such as myc-l, lymphoma arose relatively quickly (20). In a more recently developed transgenic mouse model in which mutant cyclin D1 was inserted and therefore accumulated in the nuclei, lymphoma also developed (21). These experiments, in aggregate, support the concept that the t(11;14) is insufficient and that other genetic abnormalities are required for MCL to arise. This concept is also supported by cytogenetic data on MCL in humans. Using conventional cytogenetic methods, the t(11;14) is rarely present as a sole abnormality in MCL, in less than 10% of cases (22). Comparative genomic hybridization of MCL cases also has shown gains and losses of numerous chromosomal loci in these tumors (23).
ccnd-1 encodes for a 35-kd protein, cyclin D1, that physiologically participates in controlling the cell cycle by forming complexes with either cyclin-dependent kinase (CDK)-4 or CDK6. These complexes play a role in phosphorylating (inactivating) retinoblastoma. Retinoblastoma normally acts to suppress progression of the cell cycle and therefore its inactivation promotes cell cycle progression from G1 to S phase. In addition, the transcription factor E2F-1 is released from phosphorylation, allowing its activation of genes involved in S phase. Many other molecular abnormalities also occur in MCL and appear to be involved in pathogenesis. These changes are predominantly focused on and impair three major cellular functions: cell cycle regulation, response to DNA damage, and physiologic apoptosis (24).
Clinical Findings
The median age of MCL patients is the late sixth or seventh decades in many studies (15,25,26,27). In the Non-Hodgkin’s Lymphoma Classification Project study, the median age was 63 years, with an age range of 37 to 82 years (15). Mantle cell lymphoma is uncommon under the age of 40 years and rare in patients under the age of 30 years. At the M.D. Anderson Cancer Center, the youngest patient with MCL to date was 24 years old. There is a case report of an 18-year-old girl with blastoid variant MCL (28). Systemic (B-type) symptoms occur in 30% to 50% of patients (14,24,25,26,27). Weight loss is most common, whereas night sweats and fever are less frequent and patients often have good performance status (27). Most patients with MCL present with advanced clinical-stage disease (Ann Arbor III–IV). Lymphadenopathy is very common, often generalized, and lymph nodes are typically not bulky and range in size from 2 to 5 cm (14,24,25,26,27). Extranodal sites of disease are common in MCL patients, with the gastrointestinal (GI) tract being one of the most common. Most patients with GI tract disease are asymptomatic, but a subset of patients has the syndrome known as multiple lymphomatous polyposis (MLP) that is usually associated with GI symptoms. Other common extranodal sites of involvement by MCL include the bone marrow, liver, spleen, and Waldeyer ring. Central nervous system involvement is uncommon at time of diagnosis, but can occur over the course of the disease and has been correlated with blastoid variants of MCL (29). Skin involvement is uncommon, but can occur, almost always as part of widespread disease. Because of it accessibility, we have observed MCL patients in whom the first biopsy specimen was obtained from skin on which the diagnosis of MCL was established (30). Staging workup then revealed other sites of disease.
A complete blood count can show cytopenias in patients with extensive bone marrow involvement. A leukemic phase of MCL, often with high leukocyte counts, occurs in 5% to 10% of patients but low-level (normal leukocyte count) involvement by MCL is common (25,31). Serum chemistry studies show elevated β-2–microglobulin and lactate dehydrogenase (LDH) levels in 40% to 50% of patients. Uncommonly, patients with MCL can also have a serum monoclonal paraprotein or a positive Coombs’ test (24,32).
The International Prognostic Index (IPI) of MCL patients in one study was low (score 0–1) in 23%, intermediate (score 2–3) in 54%, and high (4,5) in 23% (14). To achieve better prognostication, a MCL IPI (MIPI) has been recently proposed (33). This system uses four parameters: age, Eastern Cooperative Oncology Group (ECOG) performance status, serum LDH, and leukocyte count. Whether this new index will be widely accepted is unknown. Pathologic parameters that have been correlated with poorer prognosis include high leukocyte count, high mitotic or proliferation (Ki-67) rates, and blastoid variants. In contrast, a pathologic parameter that has been associated with a better prognosis is a mantle zone pattern (34,35). In one study from M.D. Anderson Cancer Center, patients with a pure (>90%) mantle zone pattern had a substantially better prognosis and, as a result, the authors recommended less aggressive therapy for this small subset of MCL patients (34). However, the predictive value of tumor pattern is controversial and, at most institutions, patients with the mantle zone pattern of MCL are not treated differently from other MCL patients.
Patients with MCL have a poor prognosis and are incurable using traditional cyclophosphamide, doxorubin, vincristine, and prednisone (CHOP) chemotherapy; their median overall survival is 2 to 5 years (36). Patients with blastoid variant MCL are associated with particularly short durations of response after chemotherapy and poorer overall survival (37). As a result, more aggressive and novel treatment regimens are being used for these patients. At the M.D. Anderson Cancer Center, patients with MCL are treated with an aggressive chemotherapy regimen, R-hyperCVAD (rituximab, high-dose and hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with methotrexate and cytosine arabinoside) with or without stem cell transplantation (38). To date, complete remission and survival rates are better in MCL patients treated with R-hyperCVAD than was the case for patients treated in the CHOP era. Many other novel treatment regimens are also being used or designed to treat MCL patients, with promising results (23,39,40).
Histologic Findings
Cytologic Spectrum of Mantle Cell Lymphoma
Typical (or classical) cases of MCL are cytologically composed of a monotonous population of small to medium-sized lymphoid cells with irregular nuclear contours, condensed nuclear chromatin, inconspicuous nucleoli, and scant cytoplasm (Fig. 66.1) (1,13,24,25,26,41,42,43). The nuclear contours of the neoplastic cells can vary substantially from case to case, being only slightly irregular (almost round; similar to CLL/SLL), moderately irregular (corresponding to the classical description of IDL discussed earlier), or markedly irregular (resembling the centrocytes of FL). However, in an individual neoplasm, the cells are usually very similar to each other. Centroblasts (large noncleaved cells) or immunoblasts are absent or very rare in MCL. Proliferation centers and paraimmunoblasts, as seen in CLL/SLL, are also absent in MCL.
Blastoid variants of MCL have been described and represent approximately 10% to 15% of all cases in various studies. However, the frequency of blastoid variants may be much
higher in patients who are followed for long periods of time. Norton and colleagues (43) reported that up to 40% to 50% of MCL patients developed blastoid histologic features with 20-year follow-up. In the World Health Organization (WHO) classification, two blastoid variants of MCL are recognized: classic and pleomorphic (1). Classic blastoid variant (Fig. 66.2) is composed of cells that can closely resemble lymphoblasts. These cells are small to intermediate in size and have immature chromatin and a high mitotic rate. A starry-sky pattern can be present. According to the WHO classification, this variant has at least 10 mitoses per 10 high-power (× 400) microscopic fields, and often much higher mitotic rates (20–30/high-power microscopic fields) (1). In the pleomorphic blastoid variant (Fig. 66.3), the neoplasm is composed of a heterogeneous population of cells including large cleaved or round cells that can have prominent nucleoli. Mitotic figures are also increased, and this variant can resemble large-cell lymphoma. In our experience, classic blastoid variant MCL often presents de novo. In contrast, the pleomorphic blastoid variant MCL can occur in patients with a history of MCL or in patients with another simultaneously detected site involved by typical MCL.
higher in patients who are followed for long periods of time. Norton and colleagues (43) reported that up to 40% to 50% of MCL patients developed blastoid histologic features with 20-year follow-up. In the World Health Organization (WHO) classification, two blastoid variants of MCL are recognized: classic and pleomorphic (1). Classic blastoid variant (Fig. 66.2) is composed of cells that can closely resemble lymphoblasts. These cells are small to intermediate in size and have immature chromatin and a high mitotic rate. A starry-sky pattern can be present. According to the WHO classification, this variant has at least 10 mitoses per 10 high-power (× 400) microscopic fields, and often much higher mitotic rates (20–30/high-power microscopic fields) (1). In the pleomorphic blastoid variant (Fig. 66.3), the neoplasm is composed of a heterogeneous population of cells including large cleaved or round cells that can have prominent nucleoli. Mitotic figures are also increased, and this variant can resemble large-cell lymphoma. In our experience, classic blastoid variant MCL often presents de novo. In contrast, the pleomorphic blastoid variant MCL can occur in patients with a history of MCL or in patients with another simultaneously detected site involved by typical MCL.
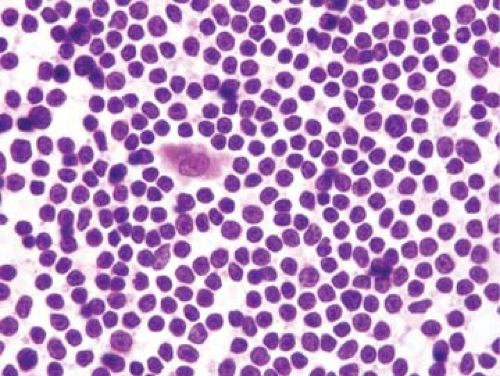 Figure 66.1. Mantle cell lymphoma involving lymph node, touch imprint. The neoplastic cells are monotonous, being small with round to slightly irregular nuclear contours. A reactive histiocyte with eosinophilic cytoplasm is also present in this field. Hematoxylin-eosin stain. |
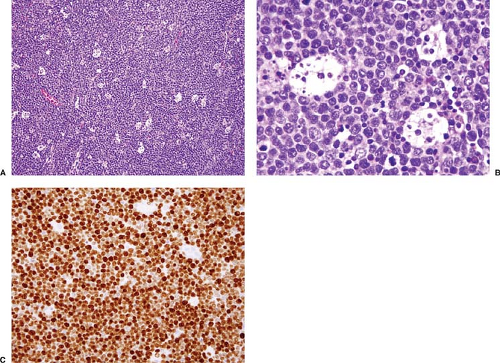 Figure 66.2. Classic blastoid variant of mantle cell lymphoma involving lymph node. A: At this magnification a starry sky pattern can be observed. B: The neoplastic cells are small with immature chromatin and a high mitotic rate. C: Immunostain for Ki-67 shows a very high proliferation rate. This neoplasm was positive for cyclin D1 (not shown) and carried the t(11;14). A,B, hematoxylin-eosin stain; C, immunohistochemistry with MIB-1 antibody and hematoxylin counterstain. |
In addition to the two cytologic extremes, typical and blastoid, other variants of MCL are recognized. Although these variants do not have clinical importance, knowledge of these variants is helpful for making the diagnosis in MCL. In the WHO classification, two other variants are specifically mentioned. A small-cell variant of MCL can be seen in which the neoplastic cells are small and round, with clumped chromatin resembling the cells of CLL/SLL. A second rare variant of MCL has foci or large areas of cells with pale cytoplasm resembling monocytoid differentiation, as can be seen in marginal zone B-cell lymphoma (Fig. 66.4).
Tiemann and colleagues (44) reviewed 304 cases of MCL and provided the relative frequency of different cytologic types of MCL. Cytologically typical (referred to as classical in this
study) neoplasms represented 87.5% of all cases of MCL, followed by blastoid (8.5%), small-cell (3.6%), and rarely mixtures of patterns including typical plus pleomorphic, and transitional cases with areas of typical and other areas with pleomorphic characteristics. In the blastoid group, the pleomorphic subtype was approximately twice as frequent as the classic subtype (44). Tiemann et al. (44) did not identify cases of MCL with marginal zone-like areas in their study.
study) neoplasms represented 87.5% of all cases of MCL, followed by blastoid (8.5%), small-cell (3.6%), and rarely mixtures of patterns including typical plus pleomorphic, and transitional cases with areas of typical and other areas with pleomorphic characteristics. In the blastoid group, the pleomorphic subtype was approximately twice as frequent as the classic subtype (44). Tiemann et al. (44) did not identify cases of MCL with marginal zone-like areas in their study.
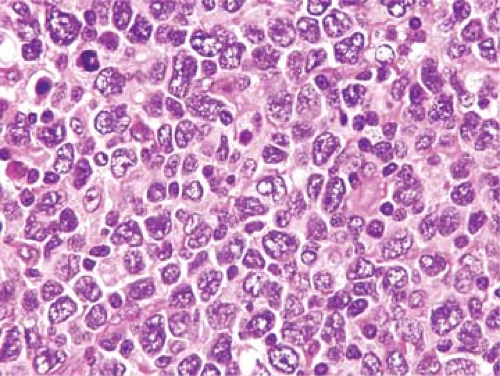 Figure 66.3. Pleomorphic blastoid variant of mantle cell lymphoma involving lymph node. This neoplasm was strongly positive for cyclin D1 (not shown) and fluorescence in situ hybridization studies showed the t(11;14). Hematoxylin-eosin stain. |
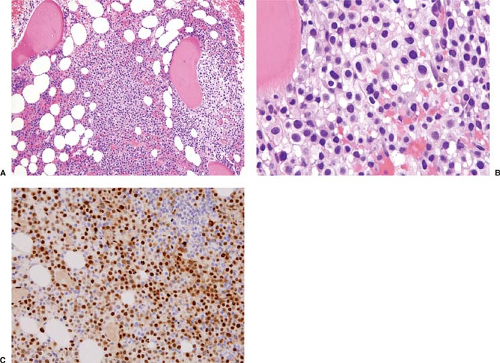 Figure 66.4. Mantle cell lymphoma with monocytoid features. A,B: The neoplasm infiltrated the bone marrow with a predominantly non-paratrabecular pattern (A) and the cells had abundant pale cytoplasm (B). C: The neoplastic cells were positive for cyclin D1. Conventional cytogenetics studies performed on bone marrow showed the t(11;14). |
Common Sites of Involvement by Mantle Cell Lymphoma
Lymph Node
Mantle cell lymphoma can exhibit one or a mixture of three histologic patterns: diffuse, nodular, or mantle-zone (1,14,25,26,27,41,42,43,44). In the diffuse pattern (Fig. 66.5), sheets of neoplastic cells efface the lymph node architecture. In the nodular pattern (Fig. 66.6), neoplastic cells are arranged in vague nodules without evidence of residual germinal centers in most of the nodules. In the mantle zone pattern (Fig. 66.7), the neoplastic cells preferentially involve and expand mantle zones surrounding small or hyperplastic benign germinal centers. In this pattern, the underlying lymph node architecture is relatively preserved. A pure mantle zone pattern, defined as greater than 90% of the entire neoplasm, may indicate a more favorable prognosis (34). A pure mantle zone pattern is rare, however, and occurs in less than 5% of cases (44). It is common for more than one pattern to be present in lymph nodes involved by MCL. These patterns of MCL may simply represent a continuum, with the mantle zone pattern present early in the course of disease. Centripedal growth by MCL then obliterates residual germinal centers and nodal architecture, evolving into nodular and then diffuse patterns. However, in patients
with widespread dissemination by MCL the mantle zone pattern rarely can be present at extranodal sites such as the GI tract. This raises the possibility that loss of the mantle zone pattern may not simply be a manifestation of increased tumor bulk, and instead may require the acquisition of additional molecular abnormalities.
with widespread dissemination by MCL the mantle zone pattern rarely can be present at extranodal sites such as the GI tract. This raises the possibility that loss of the mantle zone pattern may not simply be a manifestation of increased tumor bulk, and instead may require the acquisition of additional molecular abnormalities.
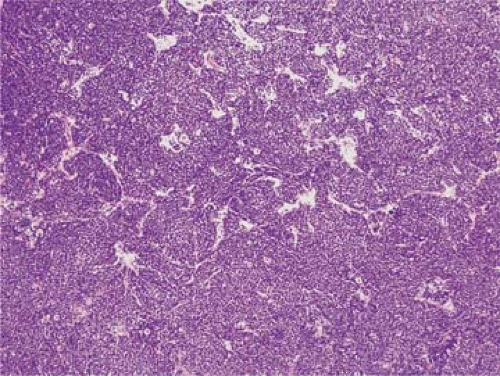 Figure 66.5. Mantle cell lymphoma, diffuse pattern, involving lymph node. Sclerotic blood vessels are also present in this field. Hematoxylin-eosin stain. |
Other histologic features are frequently observed in cases of MCL and are helpful in the recognition of this neoplasm (41,42,44). Scattered benign histiocytes with abundant eosinophilic cytoplasm (epithelioid or “pink” histiocytes) are often admixed throughout the neoplasm, in approximately two-thirds of cases of MCL (Fig. 66.8). Unlike the histiocytes observed in Burkitt lymphoma, the pink histiocytes in MCL do not have prominent apoptotic bodies in their cytoplasm, and their phagocytic capacity has been shown to be relatively inefficient. So-called “naked” germinal centers occur in approximately one-third of MCL cases. [The term “naked” germinal center is used to describe a benign germinal center completely surrounded by lymphoma, without a peripheral normal mantle zone cuff between the germinal center cells and the neoplasm (Fig. 66.8).] Last, small capillaries with hyalinized or sclerotic walls can be found in many MCLs (Fig. 66.9). It must be remembered, however, that these three histologic findings are not specific for MCL.
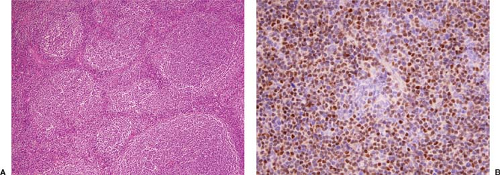 Figure 66.6. Mantle cell lymphoma, nodular pattern, involving lymph node. The poor fixation in this specimen accentuates the nodular pattern. B: The neoplastic cells express cyclin D1. A, hematoxylin-eosin stain; B, immunohistochemistry with hematoxylin counterstain. |
Gastrointestinal Tract
Approximately 10% of patients with MCL present with MLP. Although MLP was originally described as a distinctive clinical presentation of a variety of non-Hodgkin lymphomas (45), in fact, most patients with MLP have MCL (46). Patients with MLP usually come to medical attention for GI symptoms. Radiologic and endoscopic studies reveal numerous polypoid tumor masses throughout the GI tract. These polyps extend from the stomach to the rectum, but usually exclude the esophagus and anus (46,47). The extent of involvement is usually greatest and often bulky in the ileum and cecum (Fig. 66.10). Patients with MLP usually also have involvement of abdominal lymph nodes, liver, spleen, and bone marrow, but peripheral lymphadenopathy is uncommon.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


