Chapter 10 Pulmonary manifestations of systemic disease
Some systemic diseases particularly affect the lungs but are dealt with exclusively in other chapters: examples of these include sarcoidosis (p. 283), vasculitis and granulomatosis (p. 437) and Langerhans cell histiocytosis (p. 288). Other diseases often affect the lungs in isolation and are described in full elsewhere but as they also form part of systemic illnesses they also have to be referred to here: examples of these include interstitial pneumonia, which is associated with many of the systemic connective tissue disorders. Drug reactions are usually systemic, but are dealt with separately on pages 383–391.
Connective tissue diseases
The connective tissue diseases form a heterogeneous group of immunologically mediated disorders that share certain characteristics, notably inflammation of synovial and serosal membranes, connective tissue and blood vessels in various tissues. The lung is often involved in this group of diseases, probably because of its abundant connective tissue and vasculature. Pulmonary associations of connective tissue diseases include abnormalities of airways, alveoli, pulmonary blood vessels, pleura and chest wall, any of which may precede, accompany or follow the onset of the systemic disease (Table 10.1). Many of the pulmonary associations of connective tissue diseases also occur in isolation, although often accompanied by serological markers. A prime example is interstitial pulmonary fibrosis but it is notable that, in contrast to the idiopathic form of the disease, the histological pattern is more often that of non-specific interstitial pneumonia (NSIP) than usual interstitial pneumonia (UIP).1–5 Also, its progression is often slower and its prognosis correspondingly better.6–8 The few patients with connective tissue diseases who show a UIP pattern have fewer fibroblastic foci and a correspondingly better prognosis than patients with idiopathic interstitial pneumonia and UIP.9,10
The histological features of connective tissue diseases need to be separated from the effects of therapy, including drug reactions and opportunistic infections, as well as any coexistent disease processes not directly associated with the connective tissue disease. This may be difficult as, in most cases, the histological patterns found in the lung are rarely specific for a particular connective tissue disease. Detailed clinicopathological correlation is therefore essential.7
A further association of lung disease and various connective tissue disorders is seen with the pneumoconioses, particularly silicosis. There is a well-recognised association between silicosis and disorders such as systemic sclerosis and rheumatoid disease.11–13 It is doubtful whether the relationship is causal but the one seems to aggravate the other and may lead to its earlier development. For example, the presence of dust results in rheumatoid lesions in the lungs being particularly florid (see pp. 335 and 341 [Caplan’s syndrome]).
Rheumatoid disease
It was the association of pulmonary fibrosis with rheumatoid arthritis that first drew attention to the fact that this disease is not confined to the joints, and led to the use of the term ‘rheumatoid disease’ (rather than arthritis) to indicate the generalised nature of the disorder.14 It is now recognised that rheumatoid disease has many pleuropulmonary manifestations (see Table 10.1 and Box 10.1), some of which are dealt with in the appropriate chapters.
Pleurisy15–18
Pleural effusion is the commonest respiratory manifestation of rheumatoid disease. The effusion is often small and asymptomatic but it may be large and bilateral and cause pleuritic pain, fever and breathlessness. It is an exudate rather than a transudate (see p. 709). Most rheumatoid pleural effusions resolve spontaneously. Biopsy usually shows non-specific chronic inflammation of the pleura but multiple rheumatoid nodules or a linear granulomatous process showing prominent palisading of histiocytes may replace the mesothelium, in which case epithelioid histiocytes recognisable in the pleural aspirate may permit a cytological diagnosis (see Fig. 13.7, p. 714).15,19
Empyema
Patients with rheumatoid disease are prone to infections20 and occasionally a pleural effusion in a rheumatoid patient is found to be purulent. Occasionally there is pyopneumothorax,21,22 probably due to rupture into the pleural cavity of a pleural or subpleural rheumatoid nodule.
Rheumatoid nodules
Nodules identical to those found in the subcutis of patients with rheumatoid disease occasionally develop in the substance of the lung or the trachea.23–26 They may be the only manifestation of rheumatoid disease, developing before there is any arthritis or seroconversion.23 Thus, they may herald generalised disease. As they may develop in any part of the body, the occasional involvement of the major airways should occasion no surprise.25
The nodules in the lungs may be recurrent or appear first in one lung and then the other. They may be solitary or multiple and may enlarge, remain static or shrink to an insignificant scar. Resolution is usually by absorption but occasionally a nodule cavitates and discharges its necrotic contents through an airway. Erosion of both an airway and the pleura establishes a bronchopleural fistula, and perhaps pyopneumothorax.21,22 Direct involvement of the pleura by rheumatoid nodules is considered on page 714.
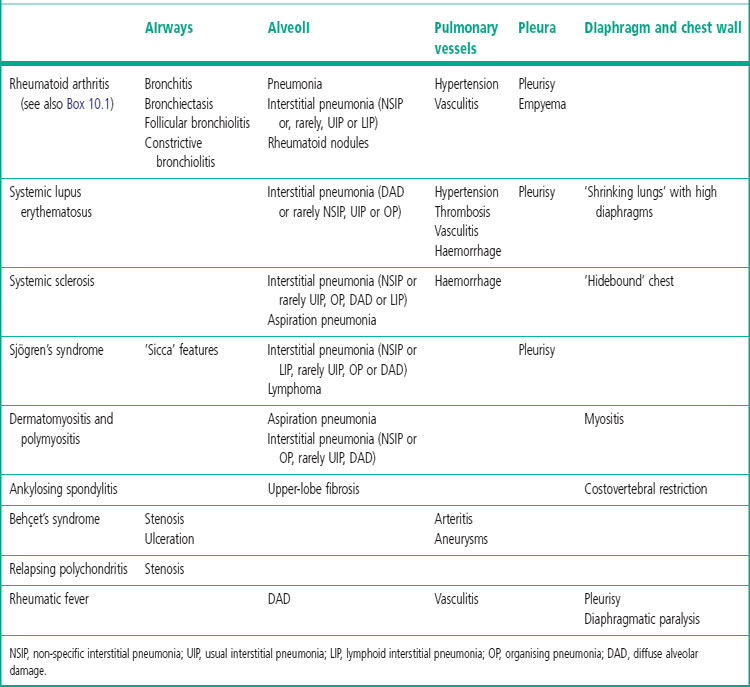
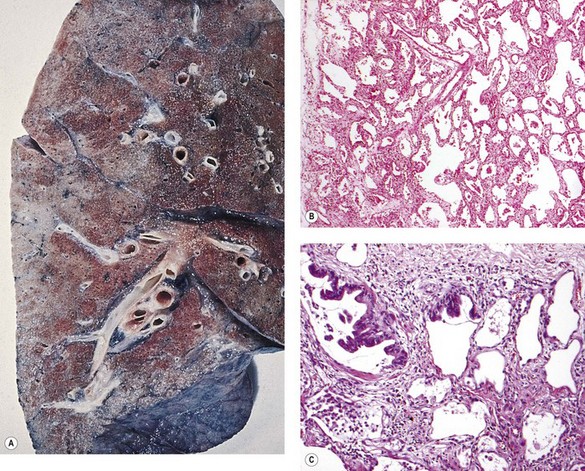
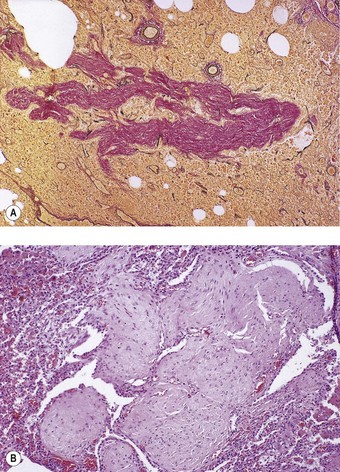
Patients with rheumatoid nodules may present with haemoptysis, or chest pain if there is pleural involvement, or they may have no symptoms. The radiographic findings may be suspected of representing malignant disease. Rheumatoid nodules have a tendency to develop in the subpleural regions of the upper and mid zones.24 They are round and typically measure up to 3 cm across but larger nodules are sometimes found, particularly when synergistic factors such as mine dust are also present (see Caplan’s syndrome, p. 341).27 As elsewhere, the nodules have a necrotic centre of finely granular eosinophilic debris surrounded by a capsule of chronic inflammatory granulation tissue (Fig. 10.1A). The boundary between dead and viable tissue is marked by a characteristic palisade of radially oriented macrophages (Fig. 10.1B). The inflammatory cells include a small number of giant cells as well as plentiful lymphocytes and plasma cells. Fungal colonisation is a rare complication of rheumatoid nodules.28
The differential diagnosis is principally from infection and Wegener’s granulomatosis (see Table 8.3.3, p. 443). The satellite granulomas seen in tuberculosis are not a feature of rheumatoid nodules, and special stains for mycobacteria and fungi give negative results. However, staining for these organisms should always be undertaken if there is any doubt about the diagnosis. Less common infective granulomas that enter the differential diagnosis include syphilis and dirofilariasis, the former requiring serological investigation and the latter step sections to identify fragments of the worm (see pp. 216 and 258 respectively). In Wegener’s granulomatosis there is also vasculitis whereas blood vessels near rheumatoid nodules show intimal fibrosis and a cuff of lymphocytes, but not a true vasculitis.
Interstitial pneumonia
Clinically overt pulmonary parenchymal disease in the form of interstitial pneumonia is reported in approximately 5% of patients with rheumatoid arthritis but the figure rises to 19% when patients without respiratory symptoms are studied by high-resolution computed tomography (HRCT).29 All seven patterns of interstitial pneumonia described in the consensus classification (see p. 264) have been described, with UIP, NSIP and organising pneumonia being especially frequent, and often seen in association with follicular bronchiolitis.5,8,10,30–34 The available data on prognosis are contradictory: some groups have reported that the prognosis of these patterns in rheumatoid disease corresponds to that seen in their idiopathic counterparts while others claim that it is better when they are associated with connective tissue disease.8–10,31,34
Apical fibrosis
Occasional patients with rheumatoid disease develop cavitating upper-lobe fibrosis similar to that described in ankylosing spondylitis (see below).35–37
Bronchial disease
Bronchitis and bronchiectasis are commoner in patients with rheumatoid arthritis than in matched controls38,39 and occasional rheumatoid patients develop bronchocentric granulomatosis.40 High resolution computed tomography (HRCT) has permitted the recognition of airway disease in a high proportion of patients with rheumatoid arthritis.41
Follicular bronchiolitis
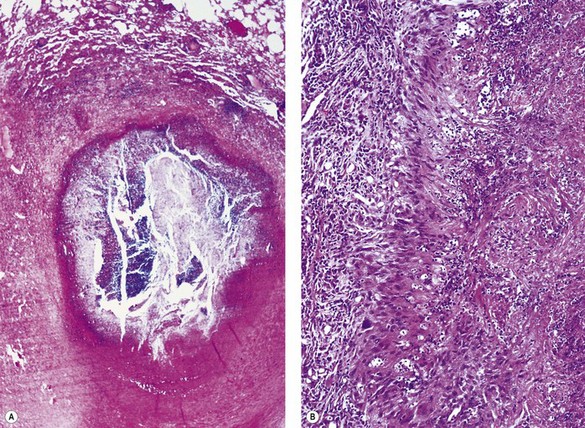
Rheumatoid disease is characterised by widespread lymphoid hyperplasia. This is best seen in lymph nodes but the bronchopulmonary lymphoid tissue is also involved in this process, resulting in the development of lymphoid follicles alongside the bronchioles, a process termed follicular bronchiolitis (Fig. 10.2).42,43 Patients complain of breathlessness with cough, fever or recurrent pneumonia. They have bilateral reticulonodular radiographic opacities and lung function tests show an obstructive, restrictive or combined defect. When follicular bronchiolitis accompanies an interstitial pneumonia it suggests that the latter is part of a systemic connective tissue disorder.10
Constrictive bronchiolitis obliterans
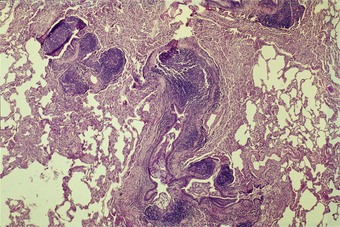
Rare patients with rheumatoid disease and other connective tissue disorders develop a bronchiolar disorder that is of considerably greater clinical consequence than follicular bronchiolitis, one that is characterised by a severe ventilatory disturbance that often proves fatal. This is the constrictive form of obliterative bronchiolitis (Fig. 10.3).44–46 The primary lesion appears to involve continuing autoimmune damage to the respiratory epithelium. This is replaced by granulation tissue, which is laid down in a circumferential pattern and gradually narrows and finally obliterates the airway lumen. It has been suggested that the process is a consequence of penicillamine47,48 or gold49 antirheumatoid therapy rather than a direct result of the connective tissue disease but as rheumatoid patients who have not received these drugs have also developed constrictive obliterative bronchiolitis this appears unlikely. Furthermore, constrictive obliterative bronchiolitis has not been reported in patients treated with penicillamine for disorders such as Wilson’s disease and primary biliary cirrhosis. Alternatively, it has been suggested that the bronchiolitis may have an infective basis. However, the obliterative process has the constrictive pattern seen when there is constant attrition of the respiratory epithelium rather than the proliferative intraluminal form of the disease seen after brief viral or chemical injury (see p. 121). It is notable that a case of obliterative bronchiolitis attributed to the domestic inhalation of a common cleansing agent concerned a patient with rheumatoid disease and that the airway obliteration was of the constrictive variety,50 suggesting that the process was the result of continuing injury rather than an isolated episode of fume inhalation.
Diffuse panbronchiolitis
This disease, which is described on page 124, is also recorded in association with rheumatoid arthritis.51,52
Pulmonary hypertension
Pulmonary hypertension is occasionally reported in rheumatoid disease. The vascular changes are described as showing advanced fibroelastic intimal proliferation that obliterates the lumen of small arteries, either in isolation53,54 or in combination with pulmonary arteritis.55 The dilatation lesions of plexogenic arteriopathy are not observed.53–55
Pulmonary vasculitis
Digital vasculitis is a common manifestation of rheumatoid disease and identical changes are occasionally encountered in the pulmonary vasculature (see Fig. 8.3.17, p. 446).55–57 It is likely that this represents a true vasculitis, probably of an immune nature, rather than the necrotising arteritis of plexogenic arteriopathy, although this is disputed.55
Pulmonary drug toxicity
Adverse pulmonary effects may be caused by drugs used in the treatment of rheumatoid disease. For example, aspirin and other non-steroidal anti-inflammatory drugs may cause asthma and eosinophilic pneumonia while gold and penicillamine may exert a cytotoxic effect resulting in diffuse alveolar damage and pulmonary fibrosis. Similarly, methotrexate may result in varying patterns of interstitial pneumonia.58
Pulmonary eosinophilia
As well as representing a reaction to the drugs used to treat rheumatoid disease, eosinophilic pneumonia may be a manifestation of the rheumatoid process itself.59
Rheumatic fever
Rheumatic fever is now rare in developed countries but remains common in many parts of the world. It involves serosal surfaces and often results in pleurisy, which is generally manifest as a sterile exudative effusion. Less commonly, there is a fibrinous alveolar exudate, organisation of which results in Masson bodies, hyaline membrane formation, chronic interstitial pneumonia or a focal necrotising process involving alveoli and pulmonary blood vessels.63–66
Systemic sclerosis
Systemic sclerosis (also known as scleroderma) is a multisystem disease that is subclassified according to the degree of skin involvement as diffuse or limited cutaneous. The former is associated with a greater degree of visceral disease and carries a worse prognosis. The limited cutaneous form is more often associated with the CREST syndrome (calcinosis cutis, Raynaud’s phenomenon, oesophageal dysfunction, sclerodactyly and telangiectasia) and has a better prognosis, although it is the form that is more often associated with pulmonary hypertension.67 Parenchymal pulmonary involvement in systemic sclerosis may take the form of interstitial pneumonia3,7,68 or diffuse alveolar haemorrhage,69,70 whilst oesophageal involvement may result in aspiration pneumonia and centriacinar fibrosis.71 Constrictive bronchiolitis is also reported72,73 and there is an increased risk of lung cancer, particularly adenocarcinoma, which is often of lepidic (bronchioloalveolar) pattern.74,75
Interstitial pneumonia
Interstitial pneumonia is the commonest pulmonary manifestation of systemic sclerosis (Fig. 10.4), the prevalence at autopsy being approximately 70%.76,77 However, many of these cases have subclinical involvement78 and the prognosis is better than in idiopathic interstitial pneumonia.7,79–81 NSIP is the commonest pattern, accounting for 55–77% of cases.2–4,8,82 The second commonest pattern is UIP but the adverse prognosis associated with this pattern in idiopathic disease is not so marked.3,4,10,82 This may be related to differences in neutrophil numbers and degranulation in the lung.83 Other histological patterns seen in systemic sclerosis include desquamative interstitial pneumonia and respiratory bronchiolitis although, it is likely that these patterns are related to smoking more than systemic sclerosis.72,84 Organising pneumonia has been reported in occasional patients,85 as has diffuse alveolar damage.33,70,86 Sarcoid granulomas have also been noted but whether they relate to systemic sclerosis or are incidental is unknown.3,87
Pulmonary hypertension
In some patients with systemic sclerosis the lung parenchyma is unremarkable but there are marked vascular changes. These patients have severe pulmonary hypertension and are mainly those with the CREST variant of systemic sclerosis (see above).67 Pathologically there is generally myxoid thickening of the wall of small pulmonary arteries, usually involving intimal fibroblasts arranged circumferentially in an ‘onion skin’ pattern (see Fig. 8.2.21, p. 431); the appearances are similar to those described in the kidney in systemic sclerosis.88,89 There is also adventitial fibrosis90 but plexiform lesions are not seen.88,91 In other patients with the CREST variant of systemic sclerosis pulmonary hypertension is caused by veno-occlusive disease.91–93
Pulmonary hypertension is also recorded in the POEMS syndrome,94,95 a rare variant of plasma cell dyscrasia with multisystem features. The name POEMS is an acronym for its principal features, polyneuropathy, organomegaly, endocrinopathy, M protein and skin changes that resemble scleroderma. It is noteworthy that in the POEMS syndrome there is overproduction of the endothelial cell-specific mitogen and potent angiogenic peptide vascular endothelial growth factor,95 which has also been reported in the plexiform lesions of other patients with pulmonary hypertension.96 Other pulmonary manifestations of the POEMS syndrome include restrictive lung disease, respiratory muscle weakness, diminished diffusing capacity and pleural effusions.97
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) is a chronic multisystem disorder associated with antinuclear autoantibodies and immune complex deposition. Women of child-bearing age are most commonly affected, the female-to-male ratio being approximately eight. Particularly common manifestations include arthropathy, skin lesions, renal disease and serositis. There are also several pleuropulmonary manifestations,98 in addition to which reactions to drugs such as methotrexate and cyclophosphamide used to treat SLE may affect the lungs. Respiratory disease is commoner in SLE than in any other connective tissue disease but the prognosis is more dependent upon renal than pleuropulmonary involvement.
Parenchymal disease
Although parenchymal disease is less common than pleural involvement, it is more serious. Acute lupus pneumonitis is characterised by diffuse alveolar damage or, less commonly, by pulmonary haemorrhage or cryptogenic organising pneumonia.99–102 Infection, which is common because of the use of immunosuppressive drugs, needs to be excluded in such acute cases.103 In survivors, healing may be by resolution but more commonly leaves the lungs permanently scarred. Chronic pulmonary injury is rarer in SLE than in rheumatoid disease and systemic sclerosis104 but both UIP and NSIP have been reported,99 and, more rarely, organising pneumonia,101,102 amyloidosis106 and lymphoid interstitial pneumonia (LIP).107 Renal involvement may lead to uraemic pulmonary oedema. Some patients with SLE are severely breathless and radiologically appear to have small lungs; the term ‘shrinking lungs’ is often applied to these cases but this is misleading as the fault lies in the respiratory muscles, which show myopathic changes, rather than in the lungs.108 Dyspnoea has been attributed to the tight skin about the chest but the cutaneous changes rarely affect respiratory function.
Pulmonary vascular disease
Pulmonary vascular disease in SLE may be secondary to systemic venous thrombosis (pulmonary thromboembolism), or widespread thrombosis may affect the pulmonary as well as the systemic circulation while diffuse alveolar haemorrhage and haemosiderosis are well-recognised complications of SLE.100,109–113 Pulmonary thrombosis, infarction and haemorrhage have been linked to the presence of circulating phospholipid antibody (also known as cardiolipin or lupus anticoagulant) in the antiphospholipid syndrome (see p. 489).114 Thrombosis may underlie the pulmonary hypertension that has been reported in association with SLE,114–119 or the pathogenesis of the hypertension may involve vasospasm, which is suggested by the frequent association of Raynaud’s phenomenon with pulmonary hypertension in SLE.115 As in systemic sclerosis, the dilatation lesions of plexogenic arteriopathy are seldom observed.116,117,119 Pulmonary veno-occlusive disease, probably due to organisation of widespread venular thrombosis, has also been described in SLE.120
Other patients with SLE have pulmonary vasculitis or capillaritis with evidence of immune complex deposition.99,121–125 The capillaritis may be complicated by diffuse alveolar haemorrhage109–112 or haemosiderosis.113 Tubuloreticular endothelial cell inclusions, once thought to be specific for SLE but later identified in AIDS and other conditions, are probably non-specific indicators of cell damage, possibly mediated by interferon.126
Polymyositis and dermatomyositis
These inflammatory disorders of skin and skeletal muscle affect women more frequently than men and have a peak onset in the sixth decade. The most common pleuropulmonary complication of these diseases is aspiration pneumonia due to a depressed cough reflex and weakness of pharyngeal and intercostal muscles and the diaphragm, which may result in ventilatory failure.129
Interstitial pneumonia
Patients with myositis may develop interstitial pneumonia, the incidence of this complication being less than 10% in patients with polymyositis130 but in the region of 41–67% in dermatomyositis.129,131,132 Patients with polymyositis and antibodies against histidyl-tRNA synthetase (Jo-1) also have a high incidence of interstitial lung disease.133–136 The commonest histological pattern is NSIP (see p. 276), which is often mixed with a component of organising pneumonia.1,134,137,138 Other patterns of interstitial lung disease that have been associated with myositis include DAD, UIP and, rarely, alveolar proteinosis.137,137a
Pulmonary vascular disease
Pulmonary vasculitis has also been reported in patients with polymyositis and the anti-Jo-1 syndrome.134,136,139 Antibodies against endothelial cells may underlie the capillaritis and resultant pulmonary haemorrhage that have been described in a few patients with dermatomyositis.139,140
Mixed connective tissue disease
Mixed connective tissue disease is characterised by overlapping features of other connective tissue disorders, notably SLE, systemic sclerosis and polymyositis, in association with high titres of an antinuclear antibody that gives a speckled pattern of immunofluorescence.142 The frequency of pulmonary involvement has ranged from 25 to 85%, the commonest form being interstitial pneumonia, followed by pleuritis, pulmonary hypertension, vasculitis and diffuse alveolar damage.33,143–147 Pulmonary hypertension is especially seen when features of systemic sclerosis are prominent but the pathological features are those of plexogenic arteriopathy rather than the myxoid intimal thickening seen in systemic sclerosis.148
Ankylosing spondylitis
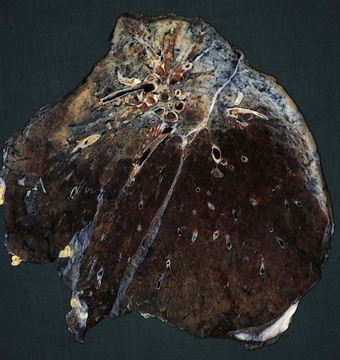
Ankylosing spondylitis typically affects the spine of young men with an HLA-B27 haplotype. Pulmonary associations include apical fibrosis. This develops in 1–2% of patients with ankylosing spondylitis, typically many years after the onset of the joint disease.149,150 Whereas the spinal lesions typically appear early in adult life, it is not until the fifth decade or so that pulmonary disease develops, often being discovered incidentally on chest radiographs. Initially, the changes may be unilateral but they have often become bilateral if cough or dyspnoea first draws attention to pulmonary involvement. As the disease progresses, cystic changes develop and the appearances resemble those of tuberculosis, but mycobacteria do not appear to play a causal role. However, like all cavities in the lungs, those of ankylosing spondylitis are liable to secondary saprophytic infection by Aspergillus species or colonisation by opportunistic mycobacteria.151
At necropsy, there is bilateral apical fibrosis (Fig. 10.5), which may be associated with bronchiectasis and bulla formation. Secondary infection may be found but generally microscopy shows only fibrosis and lymphocytic infiltration. The process is not granulomatous and the aetiology is unknown. The changes cannot be attributed to the irradiation that was formerly used to treat ankylosing spondylitis as this was confined to the spine and the pulmonary changes may develop without such therapy. Occasionally, identical changes are found in association with rheumatoid disease35 or with psoriatic spondylitis.152
Sicca syndrome and Sjögren’s syndrome
Bronchial disease
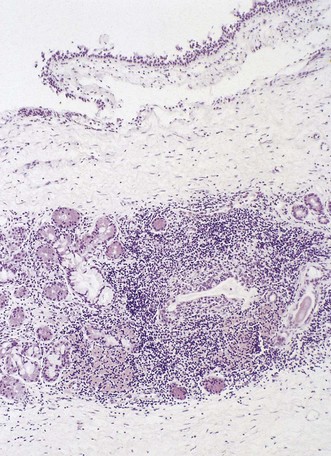
The sicca syndrome of dry eyes and dry mouth, due respectively to keratoconjunctivitis sicca and xerostomia, may be combined with one of the connective tissue disorders, usually rheumatoid disease, to form Sjögren’s syndrome. The dryness of the eyes and mouth is due to inflammatory destruction of the lacrimal and salivary glands. The same process may extend into the lower respiratory tract to destroy the tracheobronchial glands and lead to dryness of the trachea and bronchi.153–156 Airway clearance is impaired and bronchitis, bronchiectasis, bronchiolitis and pneumonia may develop. The destruction of the glands is of unknown cause. In both sicca and the full Sjögren’s syndrome, the glands show a heavy lymphoid infiltrate with destruction of the secretory acini (Fig. 10.6).157 Further extensions of the sicca syndrome are encapsulated in the term TASS syndrome (thyroiditis, Addison’s disease, Sjögren’s and sarcoidosis).158 The association with rheumatoid disease, thyroiditis and Addison’s disease suggests an autoimmune aetiology. The immunoparesis associated with human immunodeficiency virus infection is also reported in the sicca syndrome.159
Parenchymal disease
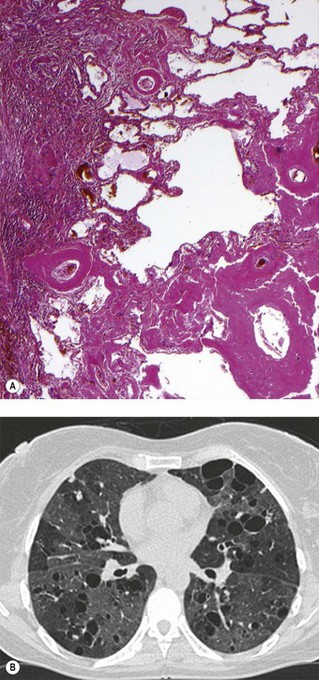
Parenchymal disease occurs in both primary and secondary Sjögren’s disease but is more common in the latter, where it often reflects the pulmonary manifestations of the associated connective tissue disease. Parenchymal disease includes amyloidosis and the formation of bullae (Fig. 10.7).160–164 Interstitial pneumonia of NSIP pattern is the commonest manifestation of primary Sjögren’s disease.165 Pulmonary hypertension, LIP, UIP and organising pneumonia are also reported.165–172
Relapsing polychondritis
Relapsing polychondritis is a rare systemic disorder affecting connective tissues with a high content of glycosaminoglycans, notably cartilage, the aorta, the sclera and cornea.176–180 It is associated with rheumatoid arthritis in about 20% of cases. Autoantibodies to cartilage and type III collagen are sometimes found181 and there may be vasculitis, glomerulonephritis, rheumatoid disease, Sjögren’s syndrome, SLE or other autoimmune disease.180,182,183
Constitutional symptoms are common while involvement of the pinnae and nasal cartilage leads to crumpled ears and a saddle-nose deformity.178,179 Involvement of the lower respiratory tract is uncommon, especially in isolation, but may result in life-threatening tracheobronchial narrowing (see p. 95).184–186
Histologically, there is erosion of the edges of the affected cartilages by a dense lymphocytic, plasma cell and giant cell infiltrate (see Fig. 3.4, p. 95). Immunoglobulins and complement components have been demonstrated at the chondrofibrous junction.187 Resolution of the inflammation, which may be spontaneous, may leave a deformed cartilage showing fibrosis and metachromasia of its ground substance.186
Behçet’s syndrome and Hughes–Stovin syndrome
Behçet’s syndrome combines recurrent orogenital ulceration with a variety of systemic disorders. A family history is sometimes obtained and the disease is associated with HLA-B51, suggesting that it has a genetic basis. Pulmonary involvement occurs in up to 8% of cases, most commonly as multiple pulmonary artery aneurysms due to a necrotising arteritis (see Fig. 8.3.19, p. 447).188 Arteries of any size may be affected. Leakage or rupture of the aneurysms causes pulmonary haemorrhage, which may be fatal. Alternatively, the arteritis may result in widespread pulmonary thrombosis.189 Other respiratory manifestations include pulmonary embolism, pleurisy, pulmonary fibrosis, bronchial ulceration and stenosis, and chyloptysis.188,190–197 The Hughes–Stovin syndrome of haemoptysis due to pulmonary artery aneurysms probably represents a limited form of Behçet’s syndrome.198–200
Cutis laxa
Cutis laxa is a rare hereditary disease of connective tissue in which the skin lacks its normal elasticity and hangs in folds like the dewlaps of a bloodhound. It may be associated with dissection of the aorta and pulmonary emphysema.201
Ehlers–Danlos syndrome
Ehlers–Danlos syndrome is another hereditary defect of connective tissue, one caused by mutation of the gene that encodes the chains of type III procollagen. There are 10 recognised subtypes, with corresponding clinical heterogeneity. The syndrome is generally characterised by soft, hyperextensible skin and hyperextensibility of the joints. Visceral abnormalities include fragility of blood vessels with consequent haemorrhage, hernias, visceral rupture, mitral valve prolapse and premature rupture of the membranes in pregnancy. Respiratory complications include tracheobronchomegaly, pulmonary laceration with subsequent haemorrhage, cystic change, fibrous pseudotumours that frequently show osseous metaplasia (Fig. 10.8), pneumothoraces and haemothorax.202–205 Occasionally some of these respiratory features are seen in isolation. Ehlers–Danlos syndrome and cutis laxa have both been associated with Mounier–Kuhn’s and Klippel–Trenaunay syndromes (see pp. 163 and 482).
Pseudoxanthoma elasticum
Pseudoxanthoma elasticum is a further generalised connective tissue disorder that has its principal manifestations in the skin but may involve the lung. As in other tissues the pulmonary elastic tissue is damaged and the site of dystrophic calcification.206
Marfan’s syndrome
Marfan’s syndrome is a further hereditary disorder of connective tissue. It is an autosomal disorder caused by mutation of the gene encoding fibrillin-1, which is a major component of the microfibrillar part of elastin.207 Dysregulation of transforming growth factor-β (TGF-β) signalling due to mutations in the TGF-β genes has also been implicated in certain patients with Marfan’s syndrome.208 Patients with Marfan’s syndrome are of a tall, thin build and have arachnodactyly, a high arched palate, pectus excavatum, scoliosis, flat feet, dislocation of the lens, aortic incompetence, dissection of the aorta and mitral valve prolapse. Pulmonary complications of the connective tissue fragility include recurrent pneumothoraces and emphysema.209–211
Vascular disease
Cardiovascular disease often leads to cardiac hypertrophy and if this is severe it may affect the lungs. For example, the enlarged heart may come to occupy a significant proportion of the thoracic space, restricting lung volume, or an enlarged left atrium may compress the oesophagus, causing swallowing difficulties and so promoting aspiration pneumonia. In infancy the bronchi are narrow and still quite pliable and are therefore liable to be compressed at certain points if the pulmonary arteries are distended, as in acyanotic congenital cardiac disease, resulting in absorption collapse (Fig. 10.9).212 Congenital heart disease may have profound effects upon the pulmonary circulation, as may failure of the left ventricle and mitral stenosis; these conditions are considered in Chapters 2 and 8.1 respectively. Pulmonary hypertension secondary to disorders of the heart and systemic vasculature is dealt with in Chapter 8.2 and vasculitis in Chapter 8.3.
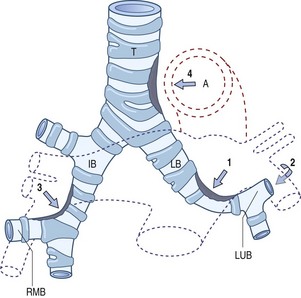
Anomalies of the great vessels compressing the trachea
The left pulmonary artery sling syndrome
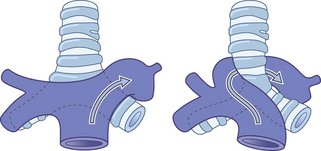
In this syndrome,213 the left pulmonary artery crosses to the right in front of the trachea, curls around it above the origin of the right main bronchus and runs to the left between the trachea and the oesophagus (Fig. 10.10). The lower trachea is liable to be compressed, causing stridor or wheeze.
Vascular rings
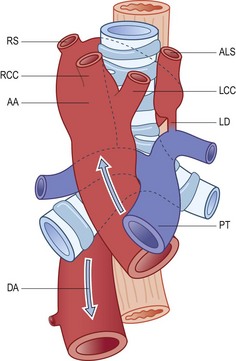
(Reproduced from Stewart et al. (1966)214 with permission from the American Journal of Roentgenology.)
It will be noted that in two of these conditions there is a right-sided aortic arch. This can be identified radiologically and in a child thought to have corticosteroid-resistant asthma is a valuable indication that the true cause of the child’s wheezing is a vascular ring compressing the trachea.215
Hereditary haemorrhagic telangiectasia (Osler–Weber–Rendu disease)
Hereditary haemorrhagic telangiectasia is an autosomal-dominant condition characterised by multiple telangiectases in the skin and mucous membranes, often resulting in distressing and sometimes life-threatening haemorrhage.216–218 Epistaxis and gastrointestinal haemorrhage are common manifestations while there may be haemoptysis, haematuria and iron-deficiency anaemia. Viscera such as the lungs, liver and brain may also be involved. The telangiectases connect arteries and veins, bypassing the normal resistance vessels so that they are subject to arterial pressure.219 They act as arteriovenous fistulae and increase cardiac output. About 15% of patients have large pulmonary arteriovenous fistulae (see p. 76).220 These permit paradoxical embolisation with the danger of infarction and abscesses in sites such as the brain.
Genetic mutations have been identified at three loci involving cell surface receptors for TGF-β. Two, which are located at 9q33-34 and 3p22 and respectively encode for endogline and ALK1, are found in families in whom pulmonary arteriovenous fistulae are common.221–223 The third mutation, which is less common, has been mapped to the long arm of chromosome 12.224 This mutation is associated with primary (plexogenic) pulmonary hypertension rather than pulmonary arteriovenous fistulae.224–226 Thus, two mechanisms underlie the development of pulmonary hypertension in hereditary haemorrhagic telangiectasia: there may be either high-output left-sided cardiac failure or primary pulmonary vasoconstriction.227
Klippel–Trenaunay syndrome
This is a congenital disorder characterised by a triad of cutaneous vascular lesions, venous abnormalities of the extremities and soft-tissue or bony hypertrophy. Venous thromboembolism is a common complication, sometimes leading to pulmonary hypertension and right heart failure.228–231 Occasionally, there are also pulmonary angiomas.
Idiopathic arterial calcification of infancy
In this condition there is calcification of the arterial internal elastic laminae, generally involving systemic vessels but also pulmonary arteries on occasion.232 Affected babies usually die in infancy and the diagnosis is often made at autopsy. Medium-sized arteries are rigid and as well as showing calcification of the internal elastic lamina they are often obliterated by secondary thrombosis and fibrosis.
Blue rubber bleb disease
Blue rubber bleb disease is characterised by cavernous haemangiomas involving the skin and gastrointestinal tract. Rarely the lesions extend into the trachea and bronchi where they are seen as discrete bluish mucosal nodules.233
Yellow-nail syndrome
This syndrome is characterised by lymphatic hypoplasia, lymphoedema, chylous effusions and yellow discoloration of the nails. It is described more fully on page 709.
Aortic fistula
Fistulas between the aorta and the tracheobronchial tree may complicate previous thoracic vascular surgery, aortic aneurysm, bacterial aortitis or radiation therapy. They often present as recurrent episodes of haemoptysis but the initial event may be a catastrophic flooding of the airways.234
Renal disease
A combination of haematuria and pulmonary haemorrhage (or haemosiderosis) is seen in a variety of pulmonary–renal syndromes (Box 10.2).
Box 10.2 Renal disease associated with pulmonary haemorrhage
GBM, glomerular basement membrane.
Microalbuminuria is a risk marker for thromboembolism, probably because it reflects generalised endothelial dysfunction.235
Renal failure
The commonest respiratory conditions encountered in renal failure are infection and pulmonary oedema. The latter is often described as ‘uraemic lung’ but is indistinguishable from pulmonary oedema of other cause, described in full on page 401. Several mechanisms may contribute to the development of pulmonary oedema in renal failure. They include fluid retention; hypertensive left ventricular failure secondary to chronic glomerulonephritis, chronic pyelonephritis and polycystic renal disease; and hypoproteinaemia, which is a prominent feature of the nephrotic syndrome.236 Cytotoxic factors may also operate.237 Pleural effusion may develop for the same reasons.
Renal failure may be complicated by secondary hyperparathyroidism, leading to absorption of bone and a secondary rise in serum calcium levels. This may be followed by metastatic calcification, in which the lungs are amongst the most commonly affected organs (see below). Pulmonary hypertension is also reported in chronic renal failure, affecting 29% of such patients in one study; its etiology is poorly understood but it appears to be unrelated to secondary hyperparathyroidism or pulmonary vascular calcification.238
The treatment of renal failure introduces further respiratory hazards. The immunosuppression involved in renal transplantation is associated with azathioprine-associated interstitial pneumonitis, opportunistic infection and posttransplantation lymphoproliferative disease, while dialysis has its own detrimental effects on lung function. Peritoneal dialysis entails a reduction in diaphragmatic excursions so that a low vital capacity and hypoxaemia are regularly seen with this form of dialysis. Haemodialysis may be accompanied by hypoxaemia, probably because the filters employed activate complement and lead to the sequestration of neutrophils in the pulmonary capillaries, as in shock (see Figs 4.20 and 4.21, p. 144).
Autosomal-dominant polycystic kidney disease
The association of bronchiectasis with this renal disorder is described on page 65.
Blood disorders
Attention here will be concentrated on the respiratory associations of diseases that are essentially haematological, although it may be noted that any lung disease liable to impair oxygenation of the blood may give rise to secondary polycythaemia while the paraneoplastic syndromes associated with lung tumours include conditions such as red cell aplasia.240 Lymphoproliferative disease is dealt with in Chapter 12.4.
Anaemia
Anaemia from any cause is liable to give rise to breathlessness because of impaired oxygen transport, even when cardiorespiratory function is normal. More specific is an association of autoimmune haemolytic anaemia with idiopathic pulmonary fibrosis.241
Hypercoaguability
Hypercoaguability of the blood gives rise to pulmonary thromboembolism, pulmonary thrombosis and pulmonary infarction, conditions that are accordingly seen in association with polycythaemia,242 thrombocythaemia,242 myelomatosis, macroglobulinaemia,243 cryoglobulinaemia,244 cryofibrinogenaemia,245 the antiphospholipid syndrome (see p. 489),246–249 the hypereosinophilic syndrome (see p. 467) and haemoglobinopathies such as sickle cell disease,250–255 spherocytosis,256 thalassaemia258,259 and paroxysmal nocturnal haemoglobinuria.260 The thrombosis and subsequent organisation may be widespread and lead to pulmonary hypertension of the pattern seen in thromboembolic disease (see p. 404).260
Sickle cell disease
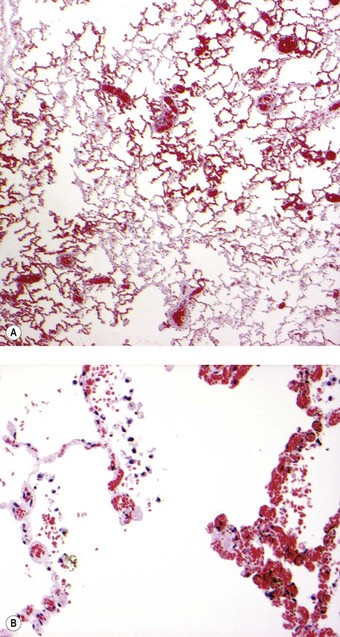
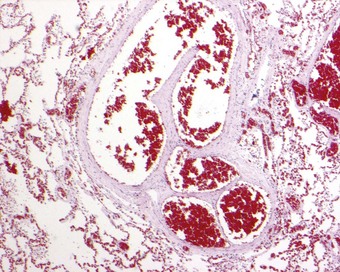
Sickle cell disease is a serious haemolytic disorder caused by homozygosity for haemoglobin S. The sickle cell trait is its milder heterozygous counterpart in which there is both haemoglobin S and A. The gene is found in Africa and Mediterranean countries. Whenever oxygen tension drops, haemoglobin S is liable to alter its molecular shape and deform the red blood cell, causing haemolysis and thrombosis. Infarction is widespread and may involve the lungs. Two major clinical forms of pulmonary involvement are recognised: an acute chest syndrome and sickle cell chronic lung disease. Acute chest syndrome is characterised by fever, chest pain and the appearance of new radiographic opacities. It represents an acute pulmonary vaso-occlusive crisis leading to acute right ventricular failure and sometimes death. Histologically, this may mimic veno-occlusive disease by causing patchy congestion through capillary sludging of the abnormal red blood cells (Fig. 10.12). Exchange transfusion has been shown to be effective.261 Chronic lung disease on the other hand is manifest as radiographic infiltrates, impaired pulmonary function and, in its most severe form, pulmonary hypertension.262–264 The pulmonary hypertension is due to pulmonary thrombosis (Fig. 10.13).250–255 In severe cases there is plexogenic arteriopathy.265,266 Veins as well as arteries thrombose and, when the venous thrombus organises, the appearances are similar to those seen in veno-occlusive disease. Rarely, pneumococcal septicaemia is also seen in sickle cell disease, possibly because of the splenic infarction that occurs in this condition. Fatal bone marrow embolism is a further occasional complication of sickle cell disease.259,267
Thalassaemia
Thalassaemia is another serious haemoglobinopathy, in this case caused by persistence of fetal haemoglobin because of a genetic inability to form adult haemoglobin A. Again there is haemolysis coupled with hypercoaguability of the blood, leading to thrombosis and infarction. Pulmonary hypertension may develop but is not as common as in sickle cell disease.258 Haemosiderin deposition is a prominent pulmonary change and may lead to interstitial fibrosis.258,259,268,269 Hypoxaemia is often noted in thalassaemia; this is possibly due to arteriovenous anastomoses in the form of ‘spider naevi’ that have been demonstrated in the periphery of the lung by postmortem injection techniques in thalassaemia patients that also have cirrhosis, which is a further complication of this disease.258 Such anastomoses are recorded in the lungs of other patients with liver disease (see below). Mixed sickle cell thalassaemia has led to fatal bone marrow embolism.259
Myeloproliferative disease
Myeloproliferative disease may extend from the bone marrow to the lung, generally involving the pulmonary interstitium. Pulmonary involvement in myelofibrosis is seen as interstitial fibrosis accompanied by foci of extramedullary haemopoiesis.270–272 In rare cases there is also air space and vascular involvement leading to pulmonary haemorrhage and pulmonary hypertension.242,273 Thus, in diseases such as polycythaemia and thrombocythaemia, pulmonary hypertension may be the result of either thromboembolism or plugging of pulmonary blood vessels by haemopoietic cells.242 Leukaemic processes also infiltrate the interstitial compartment of the lung, but generally as a terminal event274: before the lungs are directly involved they are liable to be the seat of opportunistic infection. Alveolar lipóproteinosis is also described (see p. 316). Pulmonary fibrosis is recorded in occasional patients with human T-cell lymphotropic virus type I-associated myelopathy275 and leukaemia.276 The pulmonary effects of treating leukaemia with cytotoxic drugs and by bone marrow transplantation are discussed on page 668.
Chronic granulomatous disease
Chronic granulomatous disease is characterised by opportunistic infections, both bacterial and fungal, with Staphyloccus aureus, Nocardia and Aspergillus species being particularly implicated.277,278 It is due to a genetic disorder that impairs the production of oxygen metabolites by neutrophils, eosinophils and macrophages. Over 400 gene defects are recognised, involving both mutations and inactivating recombinations. They are all recessive and may be either X-linked or autosomal.
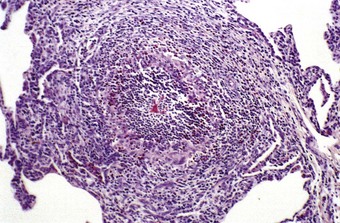
Phagocytic cells lack nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and ingested microbes are therefore not killed following phagocytosis. Abscesses develop throughout the body in the first year of life, and the lung is only one of many organs affected. Within the lung, the granulomatosis may affect the lung parenchyma or be centred on the airways.279,280 Often a balance is arrived at whereby viable microbes survive within granulomas composed of lymphocytes and yellow-brown, foamy macrophages centred on collections of neutrophils and eosinophils (Fig. 10.14).
Sea-blue histiocytosis
Sea-blue histiocytosis takes its name from the appearance of macrophages laden with cytoplasmic granules of this colour in Giemsa-stained preparations. In haematoxylin and eosin-stained sections the macrophages have an abundant pale, granular cytoplasm. Such cells are seen in Niemann–Pick disease (see below) and occasionally within the pulmonary interstitium and air spaces in a variety of acquired haematological disorders, including myelofibrosis.281
Mastocytosis
Mastocytosis is a rare disease of uncertain aetiology, characterised by infiltration and proliferation of mast cells in the skin (urticaria pigmentosa), bone marrow, gastrointestinal tract and lymph nodes. Pulmonary mast cell infiltration is particularly rare.282
IgG4-related systemic sclerosing disease
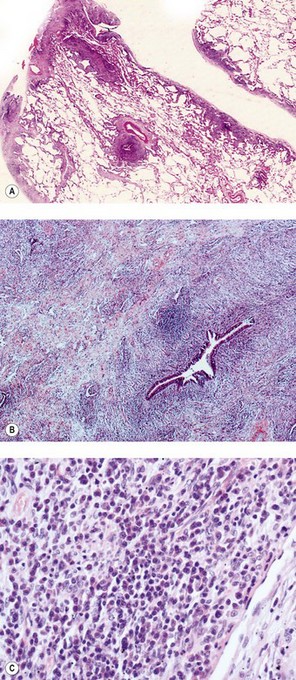
The term ‘IgG4-related systemic sclerosing disease’ has been applied to lymphoproliferative lesions found in various sites throughout the body in association with high serum levels of IgG4.283–285c The lesions contain abundant IgG4-positive cells, generally accompanying exuberant fibrous tissue, which progresses to dense hyalinosis. Idiopathic retroperitoneal fibrosis and sclerosing pancreatitis are archetypal examples. The lungs have been involved in some cases of multisystem disease286,287 and in a few instances pulmonary pseudotumours (plasma cell granulomas) have represented the sole evidence of hyper-IgG4 disease.288,289 Such patients have ranged from 42 to 78 years in age and shown an equal sex distribution. Several were asymptomatic at presentation. Imaging varies from ill-defined infiltrative lesions to nodular or multifocal opacities. Histology shows varying degrees of fibrosis with intense infiltration by lymphocytes, plasma cells, scattered neutrophils and occasional aggregates of eosinophils (Fig. 10.15A). Obliterative venulitis and obliterative arteritis (mimicking lymphomatoid granulomatosis) are common and there is sometimes a stenosing peribronchiolar infiltrate (see Fig. 10.15B, C). Immunohistochemistry shows polyclonality and many IgG4-positive plasma cells, identification of which distinguishes the condition from its principal mimics, lymphatoid granulomatosis and NSIP. Most patients have been treated surgically but it is becoming apparent that the condition responds well to systemic corticosteroids.288,290 The cases that have been classified as plasma cell granuloma or inflammatory pseudotumour need to be distinguished from inflammatory myofibroblastic tumour, a further condition to which these terms have been applied but which is now considered to be neoplastic (see p. 620). The distinction may be difficult as inflammatory myofibroblastic tumours also contain IgG4-positive plasma cells, as do conditions such as Rosai–Dorfman disease.291 The precise relationship of the IgG4-related sclerosing disease to various lung disorders requires further elucidation.
Splenectomy
Splenectomy results in lowered immunity, particularly to pneumococcal infection. Many patients who have lost their spleen have died of pneumococcal pneumonia or septicaemia and all who have undergone this operation should be offered pneumococcal immunisation. Splenectomy also appears to be a risk factor for chronic thromboembolic pulmonary hypertension.292
Alimentary tract disorders
Disorders of the upper alimentary tract
A variety of upper alimentary tract disorders may be associated with pulmonary disease, chiefly the consequence of aspiration of ingested material or of alimentary tract secretions (Box 10.3). The patient may be unaware of such aspiration. Otherwise asymptomatic gastro-oesophageal reflux has been associated with several respiratory disorders.293 Complications of aspiration include chronic cough, asthma, recurrent infections, aspiration pneumonia, abscess, empyema, bronchiectasis, exogenous lipid pneumonia, Mendelson’s syndrome (see p. 145) and chronic interstitial pneumonia and fibrosis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


