Malignant Peripheral Nerve Sheath Tumor
David R. Lucas, MD
Cyril Fisher, MD, DSc, FRCPath
Key Facts
Terminology
Sarcoma arising from a nerve or benign nerve sheath tumor or showing nerve sheath cellular differentiation
Etiology/Pathogenesis
50% associated with NF1
10% associated with radiation
Clinical Issues
Mostly adults (20-50 years)
Most (70%) arise in major nerve trunks
Local recurrence: > 40%
Metastasis: 30-60%
5-year survival: 15-34%
Microscopic Pathology
Mostly high-grade sarcomas
Spindle cell MPNST (80%)
Long fascicles of closely spaced hyperchromatic spindle cells
Small round blue cells
Pleomorphic cells
Extensive necrosis with perivascular preservation
Epithelioid MPNST (5%)
Heterologous differentiation (15%)
Ancillary Tests
S100: 50-60%
Top Differential Diagnoses
Synovial sarcoma
Cellular schwannoma
Atypical neurofibroma
Malignant melanoma
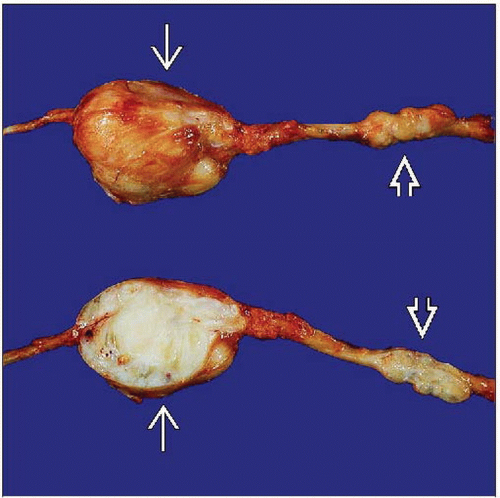 MPNSTs often arise from a major nerve trunk, such as this sciatic nerve tumor forming a fusiform, lobulated, intraneural mass  . MPNSTs can extend along the nerve to form satellite nodules . MPNSTs can extend along the nerve to form satellite nodules  . . |
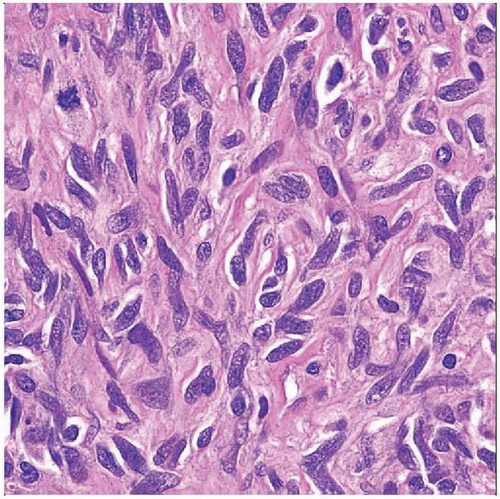 Microscopically, MPNSTs are highly variable in appearance and degree of differentiation. Well-differentiated tumors have spindle cells with tapered and wavy nuclei and indistinct cytoplasm, as shown. |
TERMINOLOGY
Abbreviations
Malignant peripheral nerve sheath tumor (MPNST)
Synonyms
Neurofibrosarcoma, malignant schwannoma, neurogenic sarcoma
Definitions
Sarcoma arising from a nerve or benign nerve sheath tumor or showing nerve sheath cellular differentiation
Diagnostic criteria
Arises from a nerve or benign nerve sheath tumor
Or shows histological evidence of nerve sheath differentiation in a NF1 patient
Or shows histological plus immunohistochemical or ultrastructural evidence of nerve sheath differentiation in non-NF1 patient
ETIOLOGY/PATHOGENESIS
Genetic Predisposition
50% associated with neurofibromatosis type 1 (NF1)
Lifetime incidence: 2-16%
40% sporadic
Environmental Exposure
10% associated with radiation
Molecular Pathogenesis
NF1 caused by germline mutation of NF1 tumor suppressor gene
Somatic loss of 2nd NF1 allele required for tumorigenesis
Malignant transformation in both NF1-associated and sporadic MPNST often involves INK4A and P53 and their downstream pathways
CLINICAL ISSUES
Epidemiology
Incidence
Rare: 5-10% of soft tissue sarcomas
Age
Mostly adults (20-50 years)
Wide age range: 10-70 years
Average age in NF1: 30 years
Average age in sporadic MPNST: 40 years
Gender
Women and men roughly equal
Site
Common sites: Thigh, buttock, trunk, upper arm, retroperitoneum, head and neck
Mostly deep-seated
Central body axis more common in NF1
Most (70%) arise in major nerve trunks
Sciatic nerve most common
Brachial plexus, sacral plexus, paraspinal nerves
Presentation
Painful mass
Neurological deficit in some
Treatment
Surgical approaches
Wide excision/resection
Amputation
Adjuvant therapy
Radiation
Drugs
Generally poor response to chemotherapy
IMAGE FINDINGS
General Features
Morphology
Large heterogeneous mass
Fusiform mass within major nerve trunk
MACROSCOPIC FEATURES
General Features
Similar to other soft tissue sarcomas
Pseudoencapsulated
Gray-tan
Firm to fleshy
Necrosis and hemorrhage common
Fusiform or eccentric mass when arising in major nerve trunk
Coexisting neurofibroma in some
Solitary or plexiform
Size
Most > 5 cm
Sometimes very massive
MICROSCOPIC PATHOLOGY
Histologic Features
Wide spectrum of cytoarchitectural patterns
Mostly high-grade sarcomas
High mitotic rate and necrosis
Only around 15% are low grade
Nerve sheath differentiation
Nuclear palisading uncommon (15%), usually focal
Tactoid differentiation with whorling or Wagner-Meissner-like features
Intraneural tumors
Plexiform architecture
Microscopic extension within nerve fascicle
Tumors arising from preexisting benign nerve sheath tumor
Neurofibroma most common, transitional areas, usually in NF1 patients
Schwannoma, ganglioneuroma, ganglioneuroblastoma, or pheochromocytoma; very rare
Diffuse sarcomatous proliferation with no evidence of nerve or nerve sheath origin
Spindle cell MPNST (80%)
Long fascicles of uniform, closely spaced, hyperchromatic spindle cells
Alternating cellular fascicles and hypocellular areas (“tapestry” or “marbled” pattern)
Storiform arrays
Small round blue cells
Pleomorphic cells
Multinucleated giant cells
Extensive necrosis with perivascular preservation
Hemangiopericytoma-like vascular pattern in some
Epithelioid MPNST (5%)
Multinodular architecture
Cords and clusters in some
Large epithelioid cells
Abundant eosinophilic cytoplasm
Large vesicular nuclei with macronucleoli
Clear cytoplasm in some
Often mixed with spindle cells
Heterologous differentiation (15%)
Osseous and osteosarcomatous
Chondroid and chondrosarcomatous
Rhabdomyosarcomatous (Triton tumor)
Angiosarcomatous
Glandular
Neuroepithelial (rosettes)
Cytologic Features
Spindle cells
Ill-defined cytoplasm
Hyperchromatic nucleus with dispersed coarse chromatin
Tapered and wavy nuclei in well-differentiated tumors
Very brisk mitotic activity in high-grade tumors
Epithelioid cells
Abundant eosinophilic or clear cytoplasm
Vesicular nucleus with prominent inclusion-like nucleolus
ANCILLARY TESTS
Immunohistochemistry
S100 protein(+) in about 60%, usually focal
Nestin(+) in 50-80%
Cytogenetics
Complex structural and numeric chromosomal abnormalities
Frequent loss of NF1 at 17q11
Frequent loss of P53 at 17q13
DIFFERENTIAL DIAGNOSIS
Monophasic or Poorly Differentiated Synovial Sarcoma
Nuclei have softer, less coarse chromatin
Usually has lower mitotic rate
TLE1(+)
MPNST rarely (2%) positive
Usually cytokeratin(+) and EMA(+)
MPNST usually negative
Usually S100(-)
t(X:18) by cytogenetics
SYT break apart by FISH
SSX-SYT fusion by RT-PCR
Cellular Schwannoma
Usually located in retroperitoneum, pelvis, posterior mediastinum
Exclusively Antoni A areas; often lacks Verocay bodies
Necrosis and mitotic figures present
Can erode/destroy bone
Lacks malignant cytological atypia
Strong, diffuse S100 staining
MPNST usually has only focal staining
Atypical Neurofibroma
Large, hyperchromatic spindle cells
Degenerated (smudged) chromatin
Low miotic rate
Usually retains cytoarchitectural features of neurofibroma
Edematous fibrillary or myxoid matrix with collagen bundles (“shredded carrots” pattern)
Malignant Melanoma
Spindle cell/sarcomatoid melanoma
May have clustered or thèque-like areas
Diffusely S100(+)
MPNST often S100(-) (50%) or with only focal staining
Usually HMB-45(-) and Melan-A(-)
Epithelioid melanoma
Amelanotic melanoma may be indistinguishable from epithelioid MPNST
Usually HMB-45(+) and Melan-A(+)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


