- Erythromycin, a commonly used macrolide antibiotic, has a broad spectrum of antibacterial activity which is similar to that of the penicillins, and so is frequently the agent of choice for patients who have penicillin allergies.
- Macrolides inhibit protein synthesis by blocking the exit of the nascent protein tunnel (in the 23S ribosomal RNA component of the large ribosome subunit) by binding to a high-affinity site inside the tunnel. The main nucleotide component of this binding pocket is adenine in prokaryotes, and the selectivity of the macrolides is due to the fact that they do not bind to guanine, which is the corresponding nucleotide in eukaryotes.
- For further information, see Zhanel et al. (2001).
Macrolide antibiotics used clinically include azithromycin, clarithromycin, erythromycin, and roxithromycin; their structures are shown in Figure 4.2.1 and their therapeutic indications are listed in Table 4.2.1.
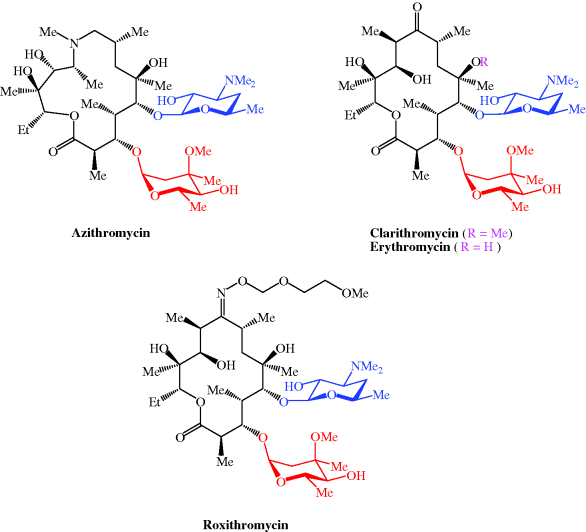
Table 4.2.1 Therapeutic indications for the macrolide antibiotics.
| Macrolide antibiotic | Indications |
| Azithromycin | Chlamydial infections, e.g. urethritis, cervicitis, community-acquired pneumonia, streptococcal pharyngitis, prevention and treatment of Mycobacterium avium complex (MAC), donovanosis, typhoid, prevention and treatment of pertussis |
| Clarithromycin | Prevention and treatment of MAC, eradication of Helicobacter pylori, prevention and treatment of pertussis |
| Erythromycin | Upper and lower RTIs, rheumatic fever prophylaxis (in penicillin allergy), legionnaires’ disease |
| Roxithromycin | Upper and lower RTIs, community-acquired pneumonia |
4.2.1 Discovery
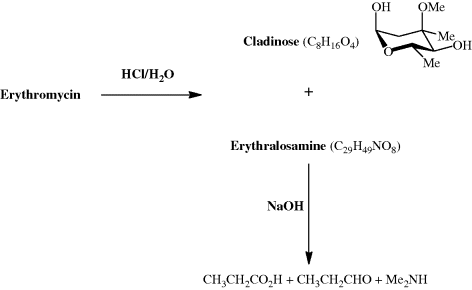
As we shall see on a number of occasions throughout this book, the discovery of the penicillins sparked worldwide interest in the antibacterial activity of microbial extracts. Samples from various locations and origins were tested in microbiology laboratories and those which exhibited antibacterial activity were selected for further study, involving the separation of the often-complex mixtures and the identification of the active component(s). The macrolide antibiotics are a product of just such a drug-discovery programme, with the agent which has been most used clinically, erythromycin, having been discovered in 1949 and introduced into the clinic in 1952. Erythromycin was discovered when researchers at Eli Lilly investigated the antibacterial activity of the metabolites of Streptomyces erythreus (now reclassified as Saccharopolyspora erythreus) isolated from a soil sample from the Iloilo region of the Philippines (for this reason, erythromycin was originally referred to as Ilotycin) (Bunch and McGuire, 1953).
Even today, with a vast array of spectroscopic techniques at our disposal, the characterisation of an unknown metabolite isolated from a natural product extract can be very challenging. How then did the research groups working, in the 1950s, on the structure of erythromycin manage to determine that it contained a 14-membered lactone (ester) ring, as well as two sugar units? (The term macrolide was coined by R. B. Woodward to denote a compound which has a macrocyclic (containing 12 or more atoms) lactone ring.) The absolute configuration of erythromycin had to wait until 1965 (Harris et al., 1965), when an X-ray crystallographic analysis established the absolute stereochemistry at each of the stereogenic centres, but the structural units had been determined a decade earlier (well before the use of NMR spectroscopy became commonplace) through extensive degradation studies in which the macrocylic ring and sugar units were converted into smaller fragments that could be identified by the comparison of their physical properties, such as melting point, with those of previously prepared and characterised authentic standards (Scheme 4.2.1) (Flynn et al., 1954). Note that, at the time this work was performed, the research group did not know the exact molecular formula for erythromycin. The products from the initial acid hydrolysis, cladinose and erythralosamine, had to be subjected to further reactions in order to determine their chemical constitution, with, for example, the dimethylamine produced by the basic hydrolysis of erythralosamine originating from the dimethylamino group in the desosamine sugar unit. You will probably also have noticed that erythromycin was introduced into the clinic before its chemical structure had been fully worked out, something which would not be possible today, given the very strict characterisation requirements of the various drug administrations around the world.
4.2.2 Synthesis
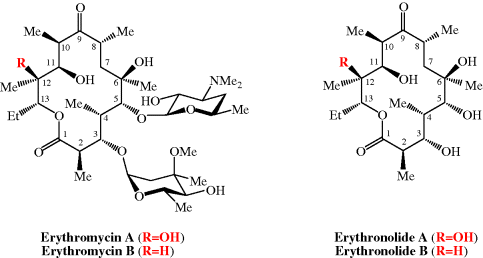
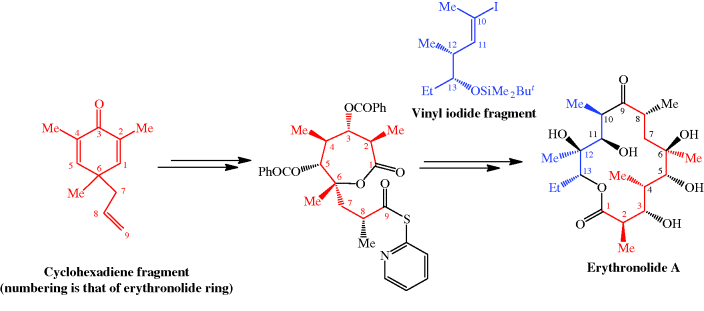
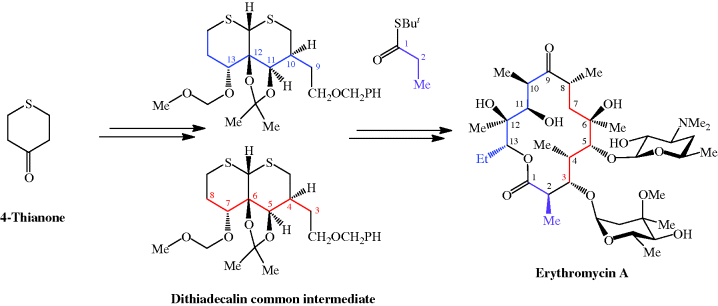
The total synthesis of the erythromycins (Figure 4.2.2) poses a supreme challenge and has attracted the attention of some of the world’s most eminent synthetic chemists, leading to many elegant examples of the total synthesis of complex natural products. The total synthesis of the erythronolide A aglycone (lacking the sugar units) was first reported by E. J. Corey (Nobel Prize in Chemistry in 1990) in a series of articles in the late 1970s (Scheme 4.2.2) (Corey et al., 1979 and references cited therein), and the total synthesis of erythromycin (known then as erythromycin A) by R. B. Woodward (Nobel Prize in Chemistry in 1965) in a series of articles in 1981, after his death (Scheme 4.2.3) (Woodward et al., 1981 and references cited therein). The Woodward synthesis is particularly elegant, as the dithiadecalin intermediate supplies both the C3-C8 and C9-C13 fragments (Scheme 4.2.3).
Scheme 4.2.2 Corey’s total synthesis of erythronolide A (38 steps from the cyclohexadiene fragment; 0.04% overall yield)
Scheme 4.2.3 Woodward’s total synthesis of erythromycin (56 steps from 4-thianone; 0.01% overall yield)
Once again, erythromycin is such a complex antibiotic that its commercial production by total synthesis will never be feasible, and it is obtained from the submerged culture of free or immobilised Saccharopolyspora erythraea (El-Enshasy et al., 2008).
We have now seen a number of examples of how very complex semi-synthetic antibiotics can be prepared through the combination of fermentation (to give the complex natural product) and chemical modification, so you will no doubt already have spotted that both clarithromycin and roxithromycin are semi-synthetic macrolide antibiotics. Clarithromycin can be obtained in a five-step synthetic procedure, from erythromycin oxime (Brunet et al., 2007), while roxithromycin can also be prepared from this oxime (Massey et al., 1970) in a single step (Scheme 4.2.4) (Gouin d’Ambrieres et al., 1982). What is not so obvious is that azithromycin is also a semi-synthetic macrolide, having originally been produced by PLIVA Pharmaceuticals from erythromycin oxime via a sequence of reactions which included the well-known Beckmann rearrangement (Djoki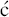 et al., 1986). For more on the synthesis of the erythromycins, see Paterson and Mansuri (1985).
et al., 1986). For more on the synthesis of the erythromycins, see Paterson and Mansuri (1985).
4.2.3 Bioavailability
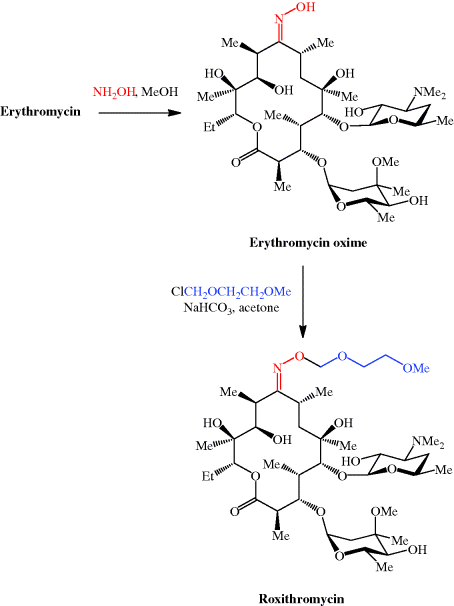
Erythromycin is sensitive to acid hydrolysis, undergoing degradation below pH 6.9, both in vitro and in vivo. Low pH also adversely affects its bioavailability, which is unpredictable (at around 15–45%) (Schentag and Ballow, 1991; Kanatani and Guglielmo, 1994) and dependent upon the exact erythromycin salt used (Billow et al., 1964; Butzler et al., 1979). Even the use of an enteric coating (which is designed to degrade and release the drug only at the higher pH of the small intestine, thereby protecting the drug from the gastric acid, pH 1–3) does not completely solve this problem, as the pH of the small intestine is 6.5–8.0, which still leads to appreciable degradation. The principal product of the acid-catalysed degradation is anhydroerythromycin (‘anhydro’ indicating the loss of water), which is formed from the hemi-acetal (Scheme 4.2.5) (Hassanzadeh et al., 2006a, 2006b). Anhydroerthryomycin has very low antibacterial activity, but its formation has further serious implications as it is a steroid 6β-hydroxylase inhibitor, as a result of its formation of a complex with cytochrome P450, and so reduces the oxidative metabolism of a range of drugs (Stupans and Sansom, 1991).
To overcome the significant problem of acid instability, and the equally important problem of extreme bitter taste, two main approaches have been taken:
- Different organic acids have been used to prepare erythromycin salts with varying aqueous solubilities, on the principle that an insoluble salt does not dissolve appreciably in saliva and so interacts less with the taste receptors, and provides a range of pharmacokinetic properties. Erythromycin stearate (Figure 4.2.3) is very insoluble in aqueous solution at <0.5 mg/mL; its low solubility at acidic pH also provides some protection from degradation, leading to slightly enhanced acid stability. Erythromycin is released from the salt in the lower GI tract when the ionised form is in equilibrium with the free amine, allowing absorption of the unionised (amine) form.
Figure 4.2.3 Structures of the salt erythromycin stearate and the prodrug erythromycin ethyl succinate (ethyl succinate prodrug moiety highlighted in blue)
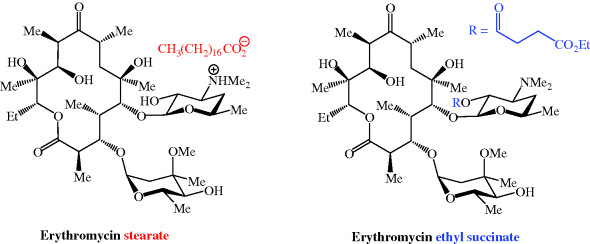
- A prodrug strategy has been used: erythromycin ethyl succinate (previously marketed as Erythroped or Eryped) consists of a double succinate ester – the carboxyl group at one end of the succinate is linked to erythromycin through its 2′-OH group and the other end is a simple ethyl ester (Figure 4.2.3) (Martin et al., 1972; Sinkula, 1974).
New salt forms of erythromycin A (Manna et al., 2004) and prodrug forms of erythromycin B, which has greater acid stability (see below), remain of interest (Hassanzadeh et al., 2007).
Pharmacokinetic evaluations of orally administered erythromycin stearate and erythromycin ethyl succinate showed that they were best absorbed, resulting in optimal bioavailability, if taken immediately before food, achieving similar maximal plasma concentrations (Cmax) of around 2.7–2.8 μg/mL at tmax 1–1.5 hours (Malmborg, 1978; Thompson et al., 1980). Taking food before administration leads to a reduction in bioavailability, and a decrease in Cmax and increase in tmax, although absorption after food was still considered to be adequate for achieving MICs and the optimal therapeutic effect. Release of erythromycin from both forms is efficient and bioequivalent to erythromycin base. The acid stability of the other macrolides is considerably better, particularly of azithromycin, which is about 10% degraded after 20 minutes at pH 2 (and 37 °C), while erythromycin will reach the same level of degradation under the same conditions in less than 4 seconds (Lode et al., 1996). The other macrolides, such as azithromycin, clarithyromycin, and the newest relative, telithromycin (which you will meet in Subsection 4.2.9), are relatively well absorbed after oral administration, achieving 37, 52, and 57% bioavailability, respectively, and their absorption is less affected by food than that of erythromycin (Zeitlinger et al., 2009). It is perhaps surprising that these molecules are so well absorbed, considering their high molecular weight, number of groups with hydrogen bonding potential, and interaction with the efflux transporter P-glycoprotein; all of which would be predicted to lead to a decrease in bioavailability. It has been suggested that their unexpectedly high absorption from the gastrointestinal (GI) tract may be the result of facilitated uptake via an as-yet-unknown mechanism (Lan et al., 2009).
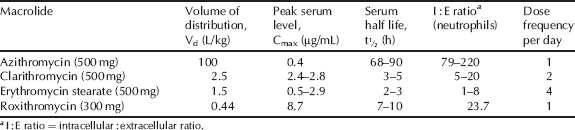
The volume of distribution of the macrolide antibiotics, alongside their low serum concentration, indicates high tissue distribution, which is supported by their intracellular accumulation and excellent activity against infection in a wide variety of tissues (Table 4.2.2). By fortunate coincidence, the macrolides accumulate in phagocytic cells, such as polymorphonuclear leukocytes (PMNLs) and macrophages, and are transported to, and subsequently released at, the site of infection, which enhances the macrolide concentration in the presence of the bacteria. Although all macrolides are taken into tissues and cells, including phagocytes, azithromycin, in particular, displays extensive selective tissue accumulation into PMNLs and macrophages and a very long tissue half-life (Amsden, 2001).
Table 4.2.2 Pharmacokinetic parameters for selected macrolide antibiotics (Amsden, 1996; Rodvold, 1999; Zhanel et al., 2001).
Clarithromycin is metabolised in the liver, primarily by oxidative N-demethylation and hydroxylation. The latter results in a major metabolite, 14(R)-hydroxyclarithromycin, which has comparable antibacterial activity to clarithromycin, is concentrated in tissue, has a longer serum half-life, and undoubtedly makes a contribution to the antibacterial activity of clarithromycin (Martin et al., 2001). Although other macrolides are subject to hepatic metabolism, the metabolites do not exhibit significant antibacterial activity (Kanatani and Guglielmo, 1994). The majority of the macrolide antibiotics are mostly eliminated through the intestinal tract, with a variable, but minor, amount excreted renally. The exception is clarithromycin and its active metabolite, 14(R)-hydroxyclarithromycin, which are primarily excreted renally in the urine.
A lot of research has been directed at improving the bioavailability and pharmacokinetic and pharmacodynamic properties of the macrolides (Zuckerman et al., 2009; Ma and Ma, 2010), through structural modifications, and this field remains of great interest (Liang et al., 2010; Sugimoto and Tanikawa, 2011).
As already mentioned, erythromycin B has greater acid stability than its better-known A form; these two molecules differ only in the substituent at C12, which is OH in erythromycin A and H in erythromycin B (Figure 4.2.2), removing the possibility of acid-catalysed cyclisation of C12-OH on to the carbonyl group at C9. Despite its similar antibacterial activity and superior acid stability, erythromycin B is not currently licensed (Tyson et al., 2011). For more on the bioavailability of the erythromycins, see Jain and Danziger (2004).
4.2.4 Mode of Action and Selectivity
During protein synthesis, the ribosome moves along the mRNA from the 5′- to the 3′-end and, once the peptide bond has formed, the non-acylated tRNA leaves the P site and the peptide-tRNA moves from the A to the P site. A new tRNA-amino acid (as specified by the mRNA codon) then enters the A site and the peptide chain grows as amino acids are added, until a stop codon is reached, when it leaves the ribosome through the nascent protein exit tunnel.
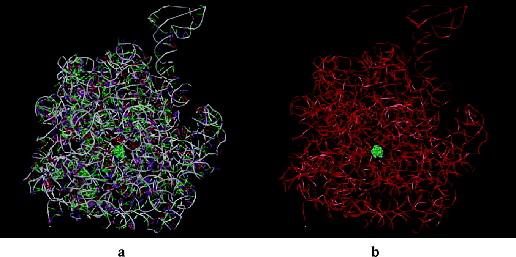
Macrolides inhibit protein synthesis by blocking the exit of this nascent protein tunnel (in the 23S ribosomal RNA component of the large ribosome subunit) by binding to a high-affinity site inside the tunnel and close to the peptidyl transferase centre, leading to the arrest of protein elongation and the dissociation of shortened peptidyl-transfer RNAs from the ribosome (Figure 4.2.4) (Schlünzen et al., 2001; Gaynor and Mankin, 2005). Although this tunnel is relatively wide, it narrows near to the peptidyl transferase centre and, as the macrolides bind at this constriction, they block the tunnel. This blockage does not affect the joining together of the first few amino acids and the polymerisation process is only halted once the growing peptide chain becomes large enough to reach the blockage.
Figure 4.2.4 Erythromycin bound inside the nascent protein exit tunnel of the ribosome from Deinococcus radiodurans; (a) showing RNA bases, and (b) with bases omitted for clarity and showing only the RNA backbone (PDB ID: 1JZY)
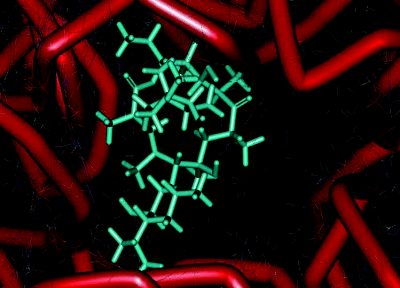
The main component of this binding pocket is nucleotide 205810 of the ribosomal RNA; in bacteria this nucleotide is adenine (A) and macrolides bind strongly to it, with the desosamine sugar (formation of hydrogen bonds between 2′-OH and A2058 and A2509; electrostatic interaction between the -N+HMe2 cation and the G2505 phosphate anion), the ring hydroxyls (hydrogen bonds between the 6, 11, and 12-OH and nucleotides), and the lactone (hydrophobic interactions) all playing key roles in the macrolide binding to this site (Figure 4.2.5) (Schlünzen et al., 2001). In eukaryotic cells, this nucleotide is guanine (G), which is larger and does not have the same favourable interactions with the 14-membered macrolides, so that the binding affinity of these antibiotics at this site is reduced, the protein tunnel in the eukaryotic ribosome is not blocked by the macrolide, and protein synthesis is unaffected. This single-nucleotide difference between the prokaryotic and eukaryotic ribosome structures is thus the basis of the selective antibacterial action of the macrolide antibiotics (Böttger et al., 2001).
Figure 4.2.5 Interaction of erythromycin with the nucleotides in its binding site in the protein exit tunnel (PDB ID: 1JZY)
4.2.5 Bacterial Resistance
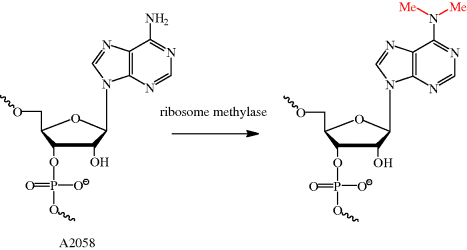
As we have seen many times already, an understanding of the mode of action of an antibiotic can also help us to understand how bacterial resistance arises. As the macrolides bind to a specific site in the ribosomal protein tunnel, it will be no surprise that bacterial resistance can arise due to alterations to this site, resulting in reduced macrolide binding affinity. For example, in streptococci, an inducible or constitutive11erm (erythromycin ribosome methylase) gene of the macrolide-lincosamide-streptogramin B (MLSB)-resistant phenotype gives rise to resistance (Roberts, 2008); in Streptococcus pneumoniae, this ribosomal methylase dimethylates a single site, A2058 (on N-6) (Scheme 4.2.6), resulting in a decreased binding affinity for erythromycin due to the reduced hydrogen bonding ability and increased size of this nucleotide. This alteration can only take place in a very small time window – it must happen during ribosome assembly, as nucleotide A2058 is buried deep within the ribosome, out of reach of the methylase once ribosome assembly is complete (Leclercq, 2002; Leclercq and Courvalin, 2002).
In Campylobacter jejuni and Campylobacter coli, which are major causes of gastroenteritis, more than one type of macrolide resistance is involved – modification of the ribosomal target for the macrolides through mutation and overexpression of a macrolide efflux pump, CmeABC (which is also involved in the intrinsic and acquired resistance of these bacteria to the quinolones and is the product of the msr gene), act in synergy to confer a high level of resistance (Payot et al., 2006). In these cases, alteration of the ribosome occurs as a result of mutations of the 2058 and 2059 nucleotides, again weakening the interaction between the macrolide and its binding site (Matsuoka et al., 1998; Vacher et al., 2005).
Finally, bacteria can develop resistance to the macrolides through the chemical inactivation of the macrolide itself. As the desosamine hydroxyl group (2′-OH) is important for antibacterial activity, deactivation of the macrolide can occur through phosphorylation (addition of phosphate) or glycosylation (addition of a sugar unit) at this position. Another chemical modification occurs in Enterobacteriaceae, which express an esterase, encoded by the ere gene, which breaks the lactone (ester) bond of the ring to give an inactive, acyclic structure (Wondrack et al., 1996).
4.2.6 Clinical Applications
4.2.6.1 Spectrum of Activity
The macrolides are broad-spectrum antibiotics and have activity against many Gram positive and some Gram negative bacteria. Erythromycin, the first commericially available macrolide, has an antibacterial spectrum similar to penicillin and is therefore often used as an alternative in patients who are allergic to pencillin. The macrolides also have the added advantage that they are active against bacteria that do not possess a cell wall (e.g. mycoplasma) – such bacteria are naturally resistant to penicillins. Macrolides are generally effective in pneumococcal, streptococcal, and Legionella infections, making them useful agents for the treatment of community-acquired pneumonia. They all have similar in vitro activity, although azithromycin appears to show superior activity against Haemophilus influenzae (Williamson and Sefton, 1993). In addition, azithromycin has activity against the opportunistic pathogens Toxoplasma gondii and Pneumocystis carinii (Araujo et al., 1988; Dunne et al., 1999).
4.2.6.2 Donovanosis
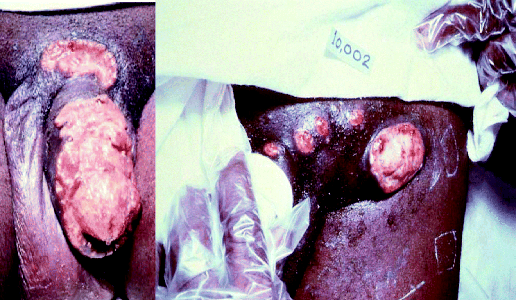
Donovanosis is a disease that can affect both men and woman and is a chronic cause of genital ulceration. Initially, the condition presents as a papule or nodule on the genital region, which then begins to ulcerate. The resulting ulcers are described as ‘beefy red’ and can bleed to the touch. If the ulcers are left untreated, they become very unsightly, causing significant psychological distress to the patient (Figure 4.2.6), and as a result patients with this condition may not seek medical advice due to embarrassment or shame. Once the diagnosis of donovanosis is made and treatment has begun, it is essential to reassure the patient about the favourable treatment outcomes (in other words, they will get better).
Figure 4.2.6 Lesions associated with donovanosis (Image courtesy of the Public Health Image Library, ID 5362, 5366. [http://phil.cdc.gov/phil/home.asp, last accessed 29th April 2011].)
Donovanosis is generally considered to be a sexually transmitted infection (STI) and is caused by the Gram negative bacterium Calymmatobacterium granulomatis, although there has been recent debate about reclassifying the organism as Klebsiella granulomatis comb nov, based on its similarity with Klebsiella species (Carter et al., 1999). The condition is relatively rare, but is more prevalent in certain parts of the world, for example, Papua New Guinea, KwaZulu Natal (part of South Africa), and parts of India and Brazil (O’Farrell, 2002).
Diagnosis of donovanosis is made by the presence of Donovan bodies (encapsulated forms of the causative organism, C. granulomatis) in tissue or biopsy samples. Once a positive diagnosis for donovanosis has been made, the current guidelines recommend azithromycin (either 1 g weekly or 500 mg daily) as first-line therapy (O’Farrell and Moi, 2010). The weekly dosing allows the azithromycin to be given by DOT,12 when deemed appropriate, thereby increasing patient compliance, and thus improving treatment outcomes (in theory, at least – as this is a rare disorder there have been no large-scale clinical trials confirming this). In cases where the patient is pregnant, erythromycin, at a dose of 500 mg four times daily, can be used, although the recent guidelines now suggest azithromycin as an alternative (O’Farrell and Moi, 2010). Treatment should generally continue until the lesions have healed, and if they are not resolved within 6 weeks further tests should be performed to exclude the possibility of cancer.
4.2.6.3 Community-Acquired Pneumonia
Community-acquired pneumonia (often just abbreviated to CAP) is a relatively common condition which is associated with significant patient morbidity and mortality. Clinical features of the condition include fever, cough, chest pain, and shortness of breath, although patients can also present with non-specific symptoms, such as confusion. The condition may be self-limiting, but antibiotics are still considered essential in the management of CAP, as their use shortens the duration of illness, reduces the risk of complications, and lowers the mortality rate.
A single pathogen is usually identified in the majority of cases (when the aetiology is confirmed – in most cases it is not), but the condition can also be caused by multiple pathogens (when this occurs the condition is known as polymicrobial CAP). S. pneumoniae is the commonest cause, but other pathogens, such as H. influenzae, Mycoplasma pneumoniae, and Legionella pneumophila, have been implicated.
When a patient presents with CAP, it is crucial to assess the severity of the condition, as this will determine what treatment they will receive and whether the condition can be managed in the community or should be referred to hospital. To assess the severity of CAP, a tool known as the CURB65 score is used. CURB65 stands for Confusion (does the patient show signs of new mental confusion?), Urea (a level raised above 7 mmol/L), Respiratory rate (raised above 30 breaths/min), Blood pressure (does the patient have a low blood pressure – either systolic (less than 90 mm Hg) or diastolic (less than 60 mm Hg?)), and finally, age (is the patient aged 65 years or more?). In cases where the patient is in hospital, the CURB65 score is used; however, in cases where the patient presents in the community (and the urea level is not easily obtainable), the simplified CRB65 score is used to assess the severity of their condition:
- If the condition is low-severity (e.g. a CURB65 score of 0 or 1, or a CRB65 score of 0), amoxicillin is recommended as monotherapy, but in cases of penicillin allergy, clarithromycin is used.
- When the condition is of moderate severity (e.g. a CURB65 score of 2), amoxicillin should be combined with clarithromycin.
- In cases of high severity (e.g. a CURB65 score of 3–5), co-amoxiclav should be combined with clarithromycin. Clarithromycin is generally preferred over erythromycin, as it causes fewer GI side effects and has an easier dosage regimen (it is given twice daily rather than four times daily).
In all of the scenarios described above, the antibiotics are given empirically (this should not be a new concept to you as we covered it in Subsection 3.2.6, when we discussed the use of trimethoprim to treat urinary tract infections in women) as they have a good spectrum of activity against a variety of organisms that cause CAP (and in particular the most common organism, S. pneumoniae). Occasionally, however, patients fail to respond to empirical antibiotic therapy and, if this does occur, cultures should be taken from the patient with a view to determining the causative organism. In cases where the organism has been identified as M. pneumoniae, clarithromycin (given either IV or orally) is recommended as first-line therapy.
One form of pneumonia that deserves a special mention is that caused by Legionella species (e.g. Legionella pneumophila) – legionnaires’ disease. This is not exclusively a form of CAP, as it can be contracted in both a hospital and the community and is rather unique as, unlike the other forms of pneumonia, it is not spread person to person – it is actually contracted by inhaling water vapour containing Legionella bacteria. These bacteria are found in natural water (e.g. rivers and lakes) and man-made water systems (e.g. water fountains), and are usually present in low numbers. Under certain conditions (such as a water temperature between 20 and 45 °C), however, the bacteria will flourish and, if ingested, can ultimately lead to infection.
There have been many sources of contamination that have led to legionnaires’ disease, with examples including air conditioning units, aerated hot tubs, water fountains, sprinkler systems, and possibly even windscreen wiper fluid in cars (Wallensten et al., 2010). Indeed, the condition gets its rather unusual name as it was first recognised in 1976 during an outbreak of pneumonia among American legionnaires attending a conference – 29 out of the reported 182 cases were fatal, illustrating how serious the condition can be (Fraser et al., 1977).
If legionnaires’ disease is suspected (clinical features which point to Legionella infection include encephalopathy and other neurological symptoms), or the patient presents with high-severity CAP, a Legionella urinary antigen test should be carried out to confirm a Legionella infection.
Erythromycin was once considered the agent of choice to treat legionnaires’ disease, but nowadays a quinolone antibiotic is recommended as first-line therapy (British Thoracic Society, 2009). In cases where a quinolone cannot be used, or is not tolerated, a macrolide (such as clarithromycin) is recommended. There have been no large-scale randomised controlled trials comparing the efficacy of macrolides and quinolones in the management of legionnaires’ disease, but results from a recent retrospective study measuring the time to clinical stability and length of hospital stay suggest that the use of levofloxacin is more favourable than macrolides – although it should be pointed out that because of low patient numbers, the results were not statistically significant (Griffin et al., 2010).
4.2.6.4 Eradication of Helicobacter pylori
The macrolides are also used (alongside other antibiotics) to eradicate H. pylori. We have already discussed H. pylori and the role it has in gastritis, peptic ulcer disease, and gastric cancer in Subsection 2.3.6, when we covered the clinical uses of the nitroimidazoles. Indeed, the macrolides (most commonly, clarithromycin) are used in combination with the nitroimidazoles and a proton pump inhibitor to eradicate H. pylori. In general, there are two common combinations used:
- PAC, which consists of a proton pump inhibitor, clarithromycin, and amoxicillin.
- PCM, which consists of a proton pump inhibitor, clarithromycin, and metronidazole.
Both regimens are thought to successfully eradicate H. pylori in around 80% of cases (Huang and Hunt, 1999; Felga et al., 2010). The optimum dose of clarithromycin used in these regimens has been open to debate, with some clinicians using clarithromycin at a dose of 250 mg twice daily and others using 500 mg twice daily. A meta-analysis which examined the doses of clarithromycin found that when the PAC regimen was used, pooled data from clinical trials showed eradication rates of 79.8% with clarithromycin 250 mg twice daily compared to 89.6% with clarithromycin 500 mg twice daily (Huang and Hunt, 1999). In this case, the 500 mg twice-daily dose of clarithromycin was statistically more effective at eradicating H. pylori. When the PCM regimen was used, however, the analysis found that doubling the dose of clarithromycin from 250 to 500 mg twice daily was not statistically more effective at eradicating H. pylori, with eradication rates of 87.4 and 88.9% reported, respectively (Huang and Hunt, 1999). The dose of clarithromycin used to eradicate H. pylori is therefore dependent on which regimen is being prescribed; if the PAC regimen is used, clarithromycin 500 mg twice daily is recommended, but if the PCM regimen is used, the lower dose of clarithromycin 250 mg twice daily is recommended.
Other macrolides can also be used to eradicate H. pylori infection, although erythromycin tends not to be used, as its use is associated with high failure rates. The newer macrolide, azithromycin, on the other hand, appears to be effective at eradicating H. pylori when used in combination with other antibiotics. One meta-analysis found that when azithromycin was used, the eradication rate was 72% (compared to 70% when it was not used) (Dong et al., 2009). Azithromycin has the added advantage that, due to its favourable pharmacokinetics, it can reach high gastric tissue concentrations, which persist for several days, meaning that it can be taken once daily for only 3 days during a 7-day treatment course. If you look again at Table 2.3.2 (which shows the common treatment regimens for eradicating H. pylori), this is clearly advantageous given the complexity and multiple dosing of the other regimens (that is, patients will find azithromycin-containing regimens easier to take, which should improve compliance with therapy). One study, however, does not share the optimism associated with the use of azithromycin for H. pylori eradication; this study found low eradication rates (at around 40%) when using azithromycin and concluded that it should not be recommended for H. pylori infection (Silva et al., 2008). This, however, serves as a good example of the importance of critically reviewing scientific literature: if we look at the doses of the agents used in the study (azithromycin 500 mg daily, omeprazole 20 mg daily, and amoxicillin 500 mg three times daily) and compare them to Table 2.3.2, you will notice that the doses and dose frequency are surprisingly different. The doses used in the study by Silva et al. are rather low compared to what is recommended, and could even be considered as ‘sub-therapeutic’. This is a possible explanation for the low eradication rates (which are not necessarily due to the fact that azithromycin has poor activity against H. pylori). The use of azithromycin for H. pylori eradication is currently not commonplace but it may have a role in the future – particularly if patient compliance becomes a problem.
4.2.6.5 Pertussis
Pertussis is an acute respiratory tract infection caused by the Gram negative bacterium Bordetella pertussis, which is highly contagious and is spread by inhaling contaminated aerosol droplets in the air (which are released through coughing or sneezing). It is characterised by bouts of severe coughing, which are often followed by a characteristic ‘whoop’ sound as the patient inhales to get their breath (yes, it really does sound like a whoop – there are plenty of examples on YouTube for you to listen to). The condition, also referred to as whooping cough, has three phases: a catarrhal phase (a dry cough may develop with signs of an upper RTI), a paroxysmal phase (the severe coughing stage often accompanied by the whoop), and a convalescent phase (when the patient starts to get better). Most people who develop pertussis make a full recovery, but complications can occasionally occur, and these include pneumonia, seizures, and, rarely, encephalopathy.
Macrolides are used first-line to treat pertussis and should be taken within 21 days of the onset of the cough (at this point, the patient is classed as still being infectious) (Health Protection Agency, 2011). They show good activity against B. pertussis and, encouragingly, there have been few reports of macrolide-resistant cases. Generally, erythromycin is the agent of choice for treatment of the infection, but as it can cause GI disturbance through interaction with motilin receptors (see Section 4.2.7), patient compliance may be an issue. When this is a problem, the newer macrolides, such as azithromycin or clarithromycin, can be used as they offer the advantage of a better side-effect profile. In addition, in infants below the age of 1 month, azithromycin is considered to be the agent of choice, as erythromycin has been associated with hypertrophic pyloric stenosis (a narrowing of the pylorus, the lower part of the stomach) in this patient group (Cooper et al., 2002).
A Cochrane systematic review found that macrolides are effective in eradicating B. pertussis, and short courses (e.g. azithromycin for 3–5 days, or clarithromycin or erythromycin for 7 days) were as efficient as longer courses of treatment (e.g. for 14 days) (Altunaiji et al., 2007). Although macrolides appear to be effective at eradicating B. pertussis, the review found they tend not to alter the clinical course of the illness, so the main reason for prescribing antibiotics in this condition is to eradicate B. pertussis and thereby prevent secondary transmission of the infection.
So far we have only considered the use of macrolides for the treatment of acute infection – they can also be used for preventative therapy when a patient has been exposed to someone with pertussis, and, interestingly, the doses for both indications are the same. Prophylaxis is recommended for all close contacts (meaning a family member, or someone living in the same household) who have been exposed to a patient who is still infectious (i.e. within 21 days of onset) and if there is a vulnerable close contact present (e.g. a newborn infant or an immunocompromised person) (Health Protection Agency, 2011).
4.2.6.6 Miscellaneous
As you will have probably gathered, there are many clinical uses of the macrolides, and we obviously cannot discuss all of them. Some of the disease states for which the macrolides are used have already been discussed, and these include:
- The prophylaxis and treatment of mycobacterium avium complex (MAC) infection when clarithromycin or azithromycin is used (see Subsection 2.2.6 for more details).
- Treatment of chlamydia with a one-off single dose of azithromycin (see Subsection 4.3.6).
- Primary prophylaxis of rheumatic fever in penicillin allergy (see Subsection 3.1.6).
- Treatment of typhoid fever caused by multiple antibacterial resistant organisms (see Subsection 2.1.6) with azithromycin.
4.2.7 Adverse Drug Reactions
There are several important adverse drug reactions associated with the macrolide antibiotics: GI disturbances and their pro-arrhythmic action, hepatotoxicity, effects on respiratory mucus transport, skin rashes, and, rarely, hearing loss.
4.2.7.1 Effects upon GI Motility
The macrolide antibiotics are associated with an increased incidence of GI disturbances: nausea, vomiting, abdominal discomfort, and diarrhoea. The incidence of these disturbances ranges from approximately 7% in patients treated with erythromycin to approximately 2.5% in patients treated with azithromycin (Hopkins, 1991). The mechanism of this adverse reaction was highlighted in the mid-1980s as being associated with motilin (Itoh et al., 1984; Zara et al., 1985), a polypeptide consisting of 22 amino acid units which stimulates the secretion of peptin. In effect, it stimulates peristalsis and accelerates gastric emptying. Motilin receptor stimulation occurs at concentrations much lower than the MICs and the use of macrolide antibiotics may lead to over-stimulation and resulting GI disturbances (Curry et al., 2001; Zatman et al., 2001).
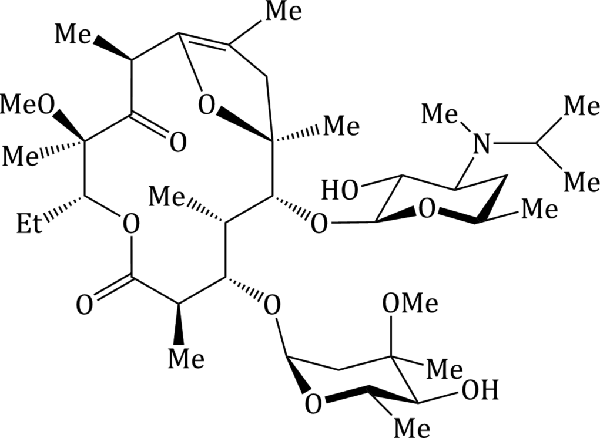
The greatest adverse effects are seen with macrolides composed of a 14-membered ring, compared to those with a 16-membered ring (Sifrim et al., 1992), and several structure–activity relationships have been conducted in an attempt to identify structural components responsible for motilin receptor activation. Several non-antibiotic, prokinetic agents (motilides) have been developed as a consequence of some of these studies, such as mitemcinal (GM-611) (Figure 4.2.7), commonly known as motilides (Stanghellini et al., 2003). Other structural activity observations are:
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


