Lymphoplasmacytic Lymphoma and Waldenstrom Macroglobulinemia
Pei Lin, MD
Key Facts
Terminology
LPL is a neoplasm of small B lymphocytes ± plasmacytoid features and plasma cells that does not meet criteria for any other small B-cell lymphoma showing plasmacytic differentiation
WM is LPL involving bone marrow associated with an IgM paraprotein of any level
Clinical Issues
Symptoms and signs of patients with WM are related to
Tissue infiltration by neoplastic cells or
Effects of elevated IgM paraprotein
WM is usually indolent; median survival: 5-10 years
Microscopic Pathology
Bone marrow involvement is constant in WM
3 subtypes of WM are recognized
Polymorphous subtype predicts poorer prognosis
Ancillary Tests
LPL and WM have 2 immunophenotypic components
B cells and plasmacytoid/plasma cells
Del6q is the most common cytogenetic aberration in WM, ˜ 40% of cases
IgH translocations are uncommon
Top Differential Diagnoses
Marginal zone lymphoma
Chronic lymphocytic leukemia/small lymphocytic lymphoma with plasmacytic differentiation
Plasma cell myeloma, small cell variant
Splenic marginal zone lymphoma
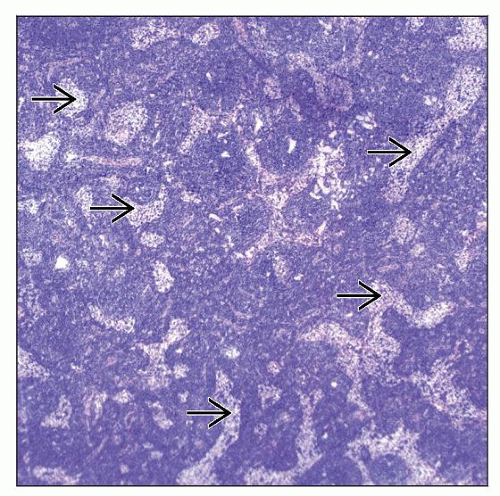 Lymphoplasmacytic lymphoma and Waldenström macroglobulinemia (LPL/WM) involving lymph node. Low-power view shows a diffuse pattern of growth with patent sinuses  that contain histiocytes. that contain histiocytes. |
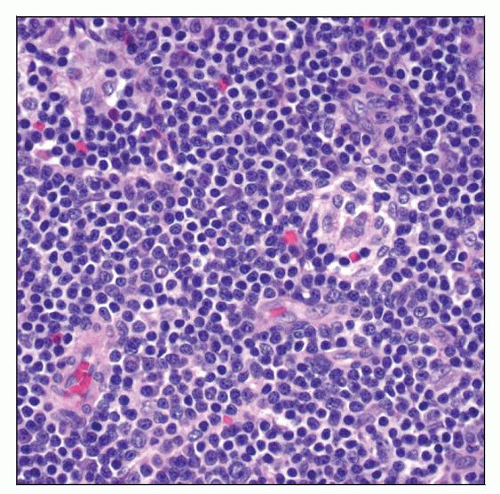 LPL/WM involving lymph node. The proliferating cells are small lymphocytes, and a subset of cells has plasmacytoid features. |
TERMINOLOGY
Abbreviations
Lymphoplasmacytic lymphoma (LPL)
Waldenström macroglobulinemia (WM)
Synonyms
Terms used in earlier lymphoma classification systems are not exact synonyms as the definition of LPL has been refined over the years
Older terms that included LPL along with other B-cell lymphomas include
Well-differentiated lymphocytic, plasmacytoid (Rappaport classification)
Immunocytoma, lymphoplasmacytic type (Kiel classification)
Malignant lymphoma, small lymphocytic, plasmacytoid (Working Formulation)
Lymphoplasmacytoid lymphoma (REAL classification)
Terms “LPL” and “WM” are often used interchangeably, but the 2 are not identical
WM represents great majority of LPL cases
Definitions
LPL is a neoplasm of small B lymphocytes, plasmacytoid lymphocytes, and plasma cells that does not meet criteria for any other type of small B-cell lymphoma showing plasmacytic differentiation
LPL is usually associated with serum monoclonal paraprotein
Usually IgM; rarely IgG or IgA
Monoclonal paraprotein is not required for diagnosis of LPL
WM is LPL involving bone marrow associated with an IgM paraprotein
No specific cutoff level of IgM is required
ETIOLOGY/PATHOGENESIS
Infectious Agents
Hepatitis C is implicated in some cases of LPL; data are controversial
Genetic Predisposition
Familial cases of WM are reported
Usually occurs in younger patients
Monoclonal gammopathy of unknown significance (MGUS) is most likely a precursor of WM
CLINICAL ISSUES
Epidemiology
Incidence
˜ 1% of non-Hodgkin lymphomas
Age
Median age: 63 to 71 years
Gender
Male to female ratio = 2:1
Ethnicity
Whites are more frequently affected than African Americans or Asians
Presentation
Relatively little data are available for patients with LPL that do not also have WM
About 25% of patients with WM are asymptomatic, so-called smoldering WM
Symptoms and signs of patients with WM are related to
Tissue infiltration by neoplastic cells or
Effects of elevated IgM paraprotein
Anemia due to bone marrow infiltration is common and causes fatigue and weakness
Bilineage cytopenia or pancytopenia can occur if infiltration by WM is extensive
Hepatomegaly occurs in ˜ 20% and splenomegaly in ˜ 15% of WM patients
WM can involve gastrointestinal tract, kidney, and other extramedullary sites
Lymphadenopathy in ˜ 15% of WM patients
Lymphadenopathy is usually mild compared with other types of non-Hodgkin lymphoma
Elevated serum IgM paraprotein levels can cause variety of symptoms
Hyperviscosity syndrome occurs in 5-15% of cases
Headache, vision disturbance (retinopathy), neurologic changes, and cardiac failure
Bing-Neel syndrome is central nervous system manifestation
Cryoglobulinemia, cold agglutinin hemolysis, autoimmune thrombocytopenia
Type II cryoglobulinemia associated with HCV may cause Raynaud phenomenon, arthralgia, skin purpura, and vasculitis
IgM-associated neuropathy is autoimmune mediated and tends to be distal, sensory, and symmetric
Deposition of IgM in skin and gastrointestinal tract causes related symptoms
Rare manifestations include amyloidosis or light-chain deposition disease
Any site: Most common in kidney, skin, or bone marrow
Laboratory Tests
IgM monoclonal protein is usually present in serum
WM
IgM paraprotein is required for diagnosis
No cutoff level for IgM in serum
LPL
IgM paraprotein not required for diagnosis
Rarely, IgA or IgG paraprotein alone or coexisting with IgM in serum
Other laboratory abnormalities well described in WM patients
Elevated erythrocyte sedimentation rate, cytopenia (usually anemia)
Elevated serum levels of LDH or β-2 microglobulin
Treatment
Treatment regimens are best defined for WM patients
Alkylating agents (chlorambucil), nucleoside analogs (cladribine or fludarabine), or single-agent rituximab is used
Autologous or allogeneic stem cell transplantation in eligible candidates
Plasmapheresis for hyperviscosity syndrome, cryoglobulinemia, neuropathy, amyloidosis, and light chain nephropathy
Corticosteroids for autoimmune hemolytic anemia or thrombocytopenia
No consensus on optimal therapy of LPL patients without WM or serum IgM paraprotein
For cases without serum paraprotein: Patients often treated with low-grade B-cell lymphoma regimens
Prognosis
WM
Usually indolent; median overall survival: 5-10 years
International prognostic scoring system designed to stratify patients into low-, intermediate-, and highrisk groups
Age > 65 years
Hemoglobin ≤ 11.5 g/dL
Platelet count ≤ 100 X 109
Serum β-2 microglobulin > 3 mg/L
Serum M protein concentration > 7 g/dL
5-year survival rates are 87%, 68%,and 36%, respectively, for low-, intermediate- and high-risk groups
Poor performance status and elevated serum LDH level also predict poorer prognosis
MACROSCOPIC FEATURES
Size
Lymph nodes are usually only modestly enlarged
MICROSCOPIC PATHOLOGY
Histologic Features
Lymph node
Involvement is usually total or subtotal, extending through capsule into perinodal adipose tissue
Pattern of growth is diffuse, but sinuses often are patent
Small residual follicles, usually in subcortical areas
Lymphoma cells are small ± plasmacytoid differentiation; monocytoid B cells can be seen
Blood vessels and sinuses may appear to be markedly dilated
Epithelioid histiocytes may be abundant or form scattered clusters
Hemosiderin or amyloid deposition
Scattered mast cells
Bone marrow involvement is constant in WM
Pattern of infiltration can be diffuse, interstitial, and nodular
3 subtypes of WM are recognized
Lymphoplasmacytoid: Usually composed of monotonous small mature lymphocytes with varying degrees of plasmacytoid differentiation
Lymphoplasmacytic: Usually composed of lymphocytes and plasmacytic (Marschalko) cells; ± Dutcher or Russell bodies
Polymorphous: Has increased (5-10%) large cells; likely represents initial progression to large B-cell lymphoma
Subtypes do not predict level of serum IgM
Peripheral blood smear
Rouleaux formation is common if IgM paraprotein level is high
Leukemic involvement (i.e., high leukocyte count) is unusual
Occasional neoplastic cells can be present
WM and LPL can be associated with so-called crystalstoring histiocytosis
Transformation of WM to diffuse large B-cell lymphoma (DLBCL) occurs in small subset of patients
WM and DLBCL components are usually of identical light chain type, suggesting clonal relationship
Serum IgM levels may paradoxically decrease
EBV is not usually implicated in transformation
Rare patients with WM can develop classical Hodgkin lymphoma (CHL)
Clonal relationship between WM and CHL is unknown
ANCILLARY TESTS
Immunohistochemistry
LPL and WM have 2 components: B cells and plasmacytoid/plasma cells
B cells
pax-5(+), CD19(+), CD20(+), CD22(+)
CD45/LCA(+), Bcl-2(+), cytoplasmic Ig(-)
CD5(-), CD10(-), Cyclin-D1(-), Bcl-6(-)
Plasmacytoid/plasma cells
CD38(+), CD138(+), CD20(-), pax-5(-/+)
Monotypic cytoplasmic Ig light chain (+), IgM(+)
Monotypic plasma cells may not be demonstrable in lymphoplasmacytoid subtype
Ki-67 typically low, p53(-), CD3(-)
Flow Cytometry
B cells
Surface IgM(+), Ig light chain (+), CD19(+), CD20(+)
Usually CD5(-), CD10(-), CD23(-)
Subset of cases may express CD5, CD10, or CD23
CD5 expression can be distinct or variable
CD10(+) cases are usually Bcl-6(-)
CD23 expression is usually dim/partial
CD11c(+/-), CD22(+/- dim), FMC-7(+/-), CD43(+/-)
CD25(-/+), CD103(-)
Plasma cells
Cytoplasmic IgM(+), Ig light chain (+)
CD19(+), CD38(+), CD138(+)
IgG type of LPL may not coexpress CD19 and CD138
Monotypic plasma cells may be only evidence of persistent disease after chemotherapy
Cytogenetics
Cytogenetic data are available for WM
Deletion of 6q in 40% of cases
Trisomy 4 in 20% of cases
Deletion of 13q in subset
Trisomy 3 is rare
Frequency of these abnormalities has not been confirmed in lymph node
IgH translocations are uncommon in WM
PAX5-IgH/t(9;14) is neither specific nor common in WM
In Situ Hybridization
EBER(-)
Array CGH
Deletions of 6q23 and 13q14
Gains of 3q13-q28, 6p, and 18q
Gains of 4q and 8q occur in 12% and 10% of WM cases, respectively
Molecular Findings in WM
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


