Lymphoplasmacytic Lymphoma
Definition
At this time, the definition of lymphoplasmacytic lymphoma (LPL) is in flux. In the World Health Organization (WHO) classification, LPL is defined primarily in the context of Waldenstrom macroglobulinemia (WM) and is named LPL/WM, although it is recognized that WM can occur in patients with other types of lymphoma (1). Lymphoplasmacytic lymphoma/Waldenstrom macroglobulinemia is defined as a neoplasm of small B lymphocytes, plasmacytoid lymphocytes, and plasma cells that usually involves the bone marrow, lymph nodes, and spleen. Clinically, patients with LPL/WM have a serum monoclonal protein, associated with hyperviscosity or cryoglobulinemia in most cases. Immunophenotypically, the neoplastic cells are usually negative for CD5 and CD10 (1). By contrast, at the Second International Workshop on WM, this disease was defined as follows: (a) presence of IgM paraprotein at any level, (b) involvement of the bone marrow by small-cell lymphoma with plasmacytoid/plasma cell differentiation, (c) intertrabecular pattern of bone marrow infiltration, and (d) an immunophenotype consistent with WM (Table 61.1) (2). The type of lymphoma involving the bone marrow is not specified as LPL in the Second International Workshop definition. Although the criteria for WM are consistent with LPL, other types of lymphoma involving bone marrow can exhibit similar morphologic and immunophenotypic features. As is apparent, the definitions of LPL/WM in the WHO classification and WM in the Second International Workshop are not synonymous.
Synonyms
Well-differentiated lymphocytic, plasmacytoid (Rappaport); immunocytoma, lymphoplasmacytic type (Kiel); malignant lymphoma, small lymphocytic, plasmacytoid (Working Formulation); lymphoplasmacytic lymphoma (World Health Organization).
Epidemiology And Etiology
In the Surveillance, Epidemiology, and End Results (SEER) data from the National Cancer Institute, LPL and WM are coded separately. Lymphoplasmacytic lymphoma and WM represent 1.7% of all non-Hodgkin lymphomas (0.7% for LPL and 1.0% for WM) and 2.3% of all B-cell non-Hodgkin lymphomas (3). The incidence rates (per 100,000 person-years) are 0.27 for LPL and 0.35 for WM (3). Men are affected more frequently than women at a ratio of almost 2 to 1, and whites are affected more frequently than African-Americans and Asians (3,4). Patients designated as having LPL or WM, depending on the study, are elderly with a median age of onset ranging from 63 to 71 years (5,6,7).
The etiology of WM is unknown. A major risk factor for developing WM is a history IgM monoclonal gammopathy of undetermined significance (MGUS). In one study by Kyle and colleagues (8), the relative risk of WM in patients with IgM-MGUS was 46 times greater than the normal population. In most patients, WM arises in a sporadic manner. However, in one study in the United States, 18.7% of patients had a family history of either WM or a plasma cell disorder (9). A familial form WM also is known, and 12 families were described in one report (10). Familial WM develops in patients at a younger age than does sporadic WM. An association between hepatitis C infection and WM or LPL has been reported, but this is controversial (11,12,13).
Clinical Findings
Patients with LPL or WM can be asymptomatic at diagnosis, in approximately 25% of patients. Asymptomatic WM also has been referred to in the literature as smoldering WM. The remaining patients manifest a variety of symptoms and signs that can be subdivided into those attributable to tissue infiltration by tumor and those attributable to the effects of monoclonal IgM secreted by the neoplastic cells.
Extensive bone marrow infiltration by tumor causes cytopenias with related symptoms (1,6,7). Anemia is the most frequent cytopenia, and is associated with fatigue and weakness. Progressive anemia is the most common indication for therapy. Other organs also can be infiltrated, but less frequently than the bone marrow. Hepatomegaly occurs in approximately 20% of patients, splenomegaly in 15%, and lymphadenopathy in 15% (7,14). Lymphadenopathy is usually mild. Other extranodal sites involved by tumor are detected in small subsets of patients and can produce local symptoms.
IgM secreted by the neoplastic cells can cause numerous pathologic consequences. Hyperviscosity syndrome is, perhaps, the best known consequence of elevated IgM in serum. However, this syndrome occurs in only a small subset of patients, from 5% to 15% at different institutions (6,7,14). Normal serum viscosity ranges from 1.4 to 1.8 centipoise (cP). When serum viscosity reaches 4 to 5 cP, symptoms can develop, although the correlation between viscosity level and symptoms is not a simple linear relationship—others factors are involved as well (6,7). The symptoms of hyperviscosity syndrome include reduced visual acuity (due to retinopathy) and ischemic manifestations in the central nervous system. The increased osmotic pressure of the serum caused by high IgM levels can lead to expanded plasma volume and high-output cardiac failure (6,7).
Serum IgM paraprotein can interact with coagulation factors or platelets, impairing their function and leading to bleeding diathesis. IgM paraprotein can become cryoglobulins, either type I (monoclonal IgM alone) or type II (combined with polyclonal IgG). Usually, type I cryoglobulins do not cause symptoms. By contrast, type II cryoglobulins can be associated with Raynaud phenomenon, arthralgia, skin purpura, and vasculitis affecting skin, kidneys, and liver. Type II cryoglobulins are also associated with hepatitis C infection (6).
IgM paraprotein also can cause symptoms due to autoantibody function (1,6,7). Approximately 15% to 30% of patients
develop peripheral neuropathy that is usually distal, sensory, and symmetric. This can be attributable to either autoantibody activity against myelin or to tissue deposition of IgM. The paraprotein can induce chronic hemolytic anemia by reacting with certain erythrocyte antigens. Patients can develop autoimmune-mediated fever, arthralgias, and skin lesions (so-called Schnitzler syndrome). Tissue deposition of IgM paraprotein also can occur. The most significant complication is primary amyloidosis as a result of tissue deposition of monoclonal Ig light chains. Tissue deposition of IgM paraprotein can also occur in the kidney, gastrointestinal tract, or skin, resulting in proteinuria, diarrhea, or skin nodules, respectively.
develop peripheral neuropathy that is usually distal, sensory, and symmetric. This can be attributable to either autoantibody activity against myelin or to tissue deposition of IgM. The paraprotein can induce chronic hemolytic anemia by reacting with certain erythrocyte antigens. Patients can develop autoimmune-mediated fever, arthralgias, and skin lesions (so-called Schnitzler syndrome). Tissue deposition of IgM paraprotein also can occur. The most significant complication is primary amyloidosis as a result of tissue deposition of monoclonal Ig light chains. Tissue deposition of IgM paraprotein can also occur in the kidney, gastrointestinal tract, or skin, resulting in proteinuria, diarrhea, or skin nodules, respectively.
TABLE 61.1 DIAGNOSTIC CRITERIA FOR WALDENSTROM MACROGLOBULINEMIA (WM) PROPOSED BY SECOND INTERNATIONAL CONFERENCE ON WM | |||||
|---|---|---|---|---|---|
| |||||
Laboratory Findings
Serum IgM Paraprotein
As stated earlier, the criteria of the Second International Workshop require that a monoclonal IgM paraprotein be present in serum (also known as M spike) to establish the diagnosis of WM (Fig. 61.1). As WM is defined, most patients with WM are presumed to have LPL. However, the presence of a serum IgM paraprotein is not required for the diagnosis of LPL in the WHO classification (1). Furthermore, the presence of serum IgM paraprotein is recognized by the WHO classification as not being specific for LPL (1). Thus, some degree of inconsistency must be addressed by participants in future International Workshops and the next iteration of the WHO classification.
The lack of specificity of a serum IgM paraprotein for a specific type of lymphoma has been known for many years (15). However, a study from the M. D. Anderson Cancer Center of 382 patients with lymphomas classified according to the WHO classification and associated with a serum IgM paraprotein illustrates this point (16). In this study, 225 (58.9%) patients had LPL/WM. The remainder of the patients had WM with other types of lymphoma; most commonly chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL; 20.2%) and, less frequently, marginal zone B-cell lymphomas (7.1%), follicular lymphoma (4.7%), large B-cell lymphoma (3.1%), mantle cell lymphoma (2.9%), and angioimmunoblastic T-cell lymphoma (1%).
It is true that the level of serum paraprotein is generally higher in patients with LPL/WM compared with patients with other types of lymphoma (1). This led others to suggest a minimum cutoff that might help separate WM patients with LPL from other types of lymphoma. For example, an arbitrary cutoff of 3 g/dL was suggested by investigators at the Mayo Clinic (17). More recently, cutoffs were discarded by the participants of the International Workshop (2). The study from the M.D. Anderson Cancer Center supports this decision (16). In the group of 225 patients with LPL/WM, the median serum paraprotein level was 2.2 g/dL, but levels ranged from 0.2 to 10.9 g/dL. Although serum paraprotein levels of greater than 3 g/dL occurred almost exclusively in LPL/WM, approximately half of LPL/WM patients had a serum IgM level of less than 3 g/dL.
Levels of serum paraprotein also correlate with the degree of symptoms. In one study, the median monoclonal IgM level was 1.8 g/dL (range 0.1 to 7.0) for asymptomatic patients with WM versus 2.2 g/dL (range, 0.2 to 3.6) for patients with symptoms (18). The substantial overlap in the range of paraprotein levels, however, precludes using serum IgM paraprotein levels to predict clinical status. In an individual patient, assessing serum paraprotein levels over time is an effective means of monitoring a patient’s disease.
Levels of serum paraprotein also loosely correlate with the degree of rouleaux observed in peripheral blood or bone marrow aspirate smears. The presence of rouleaux is best assessed in thin areas of the smear where the erythrocytes are evenly spread out and almost touching each other. When rouleaux are present, the erythrocytes form “stacks of coins,” as has been described by others (Fig. 61.2).
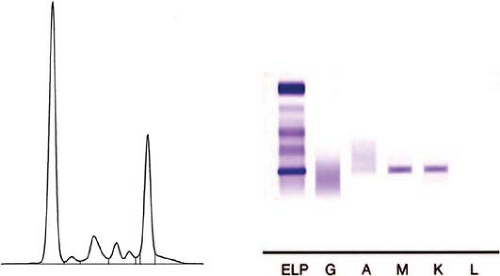 Figure 61.1. Monoclonal IgM kappa spike in the serum of a patient with both lymphoplasmacytic lymphoma (LPL) and Waldenström macroglobulinemia (WM). Left. Tracing of serum proteins showing a prominent spike in the IgM region. Right. Immunofixation showing sharp bands reactive with anti-IgM and anti-kappa. ELP, electropherogram. (Figure courtesy of Beverly C. Handy, M.D., M.D. Anderson Cancer Center) |
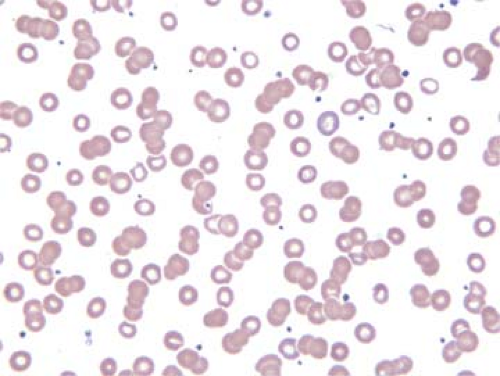 Figure 61.2. Peripheral blood smear showing rouleaux in a patient with LPL/WM. Wright-Giemsa stain. |
Other Abnormalities
Complete blood counts showing one or more cytopenias are common in patients with WM, and cytopenias are an important reason to initiate therapy (6,7). Prognostic models are based, in part, on the extent of cytopenias. Examination of the peripheral blood smear usually shows normochromic, normocytic anemia; rouleaux may be identified. The presence of rouleaux correlates with the level of paraprotein and serum viscosity. Approximately half of patients with WM also can have mild lymphocytosis with circulating small lymphocytes or lymphoplasmacytoid cells (19,20,21,22). Coagulation tests may be abnormally prolonged; this is attributed to paraprotein binding with clotting factors. The erythrocyte sedimentation rate is often high. Serum chemical analysis can show an elevated β-2 microglobulin or lactate dehydrogenase (LDH) levels in a subset of patients, and β-2 microglobulin is included in prognostic models. Hypercalcemia is uncommon in patients with LPL or WM.
Histopathology
Bone Marrow
As defined by the Workshop, the bone marrow is consistently involved in patients with WM (2). The WHO classification also recognizes that bone marrow involvement is very common in LPL/WM (1). From this point forward (for simplicity) we will use the term LPL/WM for neoplasms that meet the criteria for both LPL/WM and WM as defined in the WHO and Workshop, respectively.
In bone marrow biopsy specimens, all possible patterns of involvement by LPL/WM can occur, and the frequency of these patterns varies according to different studies. In one study by Owen and colleagues (23), the pattern was usually intertrabecular, either diffuse, interstitial, or a mixture of both patterns (Fig. 61.3). Nodular or paratrabecular patterns occurred uncommonly in LPL/WM, in approximately 5% of patients for each (23). By contrast, in a study from the Mayo Clinic,
nodular or interstitial (or both) patterns were most common, and a paratrabecular component was observed in 42% of cases (17). Cytologically, the neoplastic cells exhibit a spectrum of features resembling small lymphocytes (Fig. 61.4A), plasmacytoid lymphocytes, or mature plasma cells (Marschalko type) (Fig. 61.4B); a mixture of cell types is often present (1,16,17). The neoplastic cells may have nuclear pseudoinclusions (Dutcher bodies) or contain cytoplasmic globules (Russell bodies) of Ig (24). Mast cells are increased in a large subset of LPL/WM cases. Amyloid deposits can be seen in a small subset of LPL/WM cases (17) (Fig. 61.5).
nodular or interstitial (or both) patterns were most common, and a paratrabecular component was observed in 42% of cases (17). Cytologically, the neoplastic cells exhibit a spectrum of features resembling small lymphocytes (Fig. 61.4A), plasmacytoid lymphocytes, or mature plasma cells (Marschalko type) (Fig. 61.4B); a mixture of cell types is often present (1,16,17). The neoplastic cells may have nuclear pseudoinclusions (Dutcher bodies) or contain cytoplasmic globules (Russell bodies) of Ig (24). Mast cells are increased in a large subset of LPL/WM cases. Amyloid deposits can be seen in a small subset of LPL/WM cases (17) (Fig. 61.5).
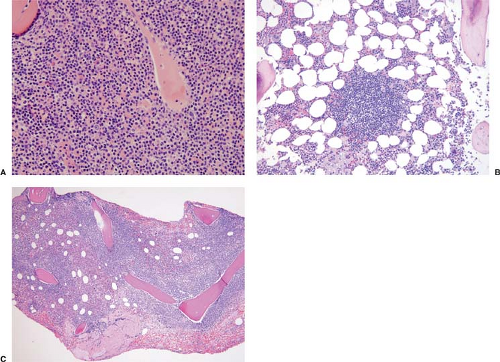 Figure 61.3. Patterns of involvement in bone marrow biopsy specimens of patients with LPL/WM. A: Diffuse pattern. B: Nodular pattern. C: Paratrabecular pattern. Hematoxylin-eosin stains. |
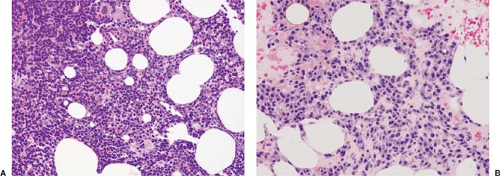 Figure 61.4. Cytologic features of two cases of LPL/WM involving bone marrow biopsy specimens. A: This neoplasm is composed mostly of small lymphocytes, the so-called lymphoplasmacytoid variant. B: This neoplasm is composed mostly of small, mature-appearing plasma cells, the so-called lymphoplasmacytic variant. Hematoxylin-eosin stains. |
In bone marrow aspirate smears and touch imprints, the spectrum of cytologic features in LPL/WM is more easily appreciated (Fig. 61.6). Mast cells are readily identified because of their metachromatic granules in Wright-Giemsa stains (Fig. 61.6A). The criteria for small lymphocytes and plasma cells are well standardized. A plasmacytoid lymphocyte is a transitional form between a small lymphocyte and a mature plasma cell. We define a plasmacytoid lymphocyte as a cell with an eccentrically located nucleus slightly larger than that of a lymphocyte and moderately abundant cytoplasm and/or cartwheel-like (plasma cell–like) chromatin (25). The typical paranuclear clear region (hof) of a plasma cell is usually not present.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


