KEY CONCEPTS
![]() Patients with Hodgkin lymphoma present with a painless, rubbery lymph node, which most commonly resides in the neck (cervical or supraclavicular nodes).
Patients with Hodgkin lymphoma present with a painless, rubbery lymph node, which most commonly resides in the neck (cervical or supraclavicular nodes).
![]() Patients with early stage Hodgkin lymphoma should be treated with combination chemotherapy with or without involved-field radiation.
Patients with early stage Hodgkin lymphoma should be treated with combination chemotherapy with or without involved-field radiation.
![]() Combination chemotherapy with doxorubicin (Adriamycin), bleomycin, vinblastine, and dacarbazine (ABVD) is the primary treatment for patients with advanced-stage Hodgkin lymphoma. Patients with advanced unfavorable disease may be treated with more aggressive regimens that have greater activity, but are associated with a higher risk of secondary malignancies.
Combination chemotherapy with doxorubicin (Adriamycin), bleomycin, vinblastine, and dacarbazine (ABVD) is the primary treatment for patients with advanced-stage Hodgkin lymphoma. Patients with advanced unfavorable disease may be treated with more aggressive regimens that have greater activity, but are associated with a higher risk of secondary malignancies.
![]() Some patients with Hodgkin lymphoma will be refractory to initial therapy or will have a recurrence following a complete remission. Response to salvage therapy depends on the extent and site of recurrence, previous therapy, and duration of initial remission. High-dose chemotherapy and autologous hematopoietic stem cell transplantation should be considered in patients with refractory or relapsed disease.
Some patients with Hodgkin lymphoma will be refractory to initial therapy or will have a recurrence following a complete remission. Response to salvage therapy depends on the extent and site of recurrence, previous therapy, and duration of initial remission. High-dose chemotherapy and autologous hematopoietic stem cell transplantation should be considered in patients with refractory or relapsed disease.
![]() The current classification system for non-Hodgkin lymphoma is the World Health Organization classification system, which is based on the principle that non-Hodgkin lymphomas can be classified into specific disease entities, defined by a combination of morphology, immunophenotype, genetic features, and clinical features.
The current classification system for non-Hodgkin lymphoma is the World Health Organization classification system, which is based on the principle that non-Hodgkin lymphomas can be classified into specific disease entities, defined by a combination of morphology, immunophenotype, genetic features, and clinical features.
![]() As compared with Hodgkin lymphoma, the clinical presentation of non-Hodgkin lymphoma is more variable because of disease heterogeneity and more frequent extranodal involvement.
As compared with Hodgkin lymphoma, the clinical presentation of non-Hodgkin lymphoma is more variable because of disease heterogeneity and more frequent extranodal involvement.
![]() The Ann Arbor staging system correlates poorly with prognosis in non-Hodgkin lymphoma because the disease does not spread through contiguous lymph nodes and often involves extranodal sites.
The Ann Arbor staging system correlates poorly with prognosis in non-Hodgkin lymphoma because the disease does not spread through contiguous lymph nodes and often involves extranodal sites.
![]() Several prognostic models have been developed to estimate prognosis in patients with non-Hodgkin lymphoma. The International Prognostic Index (IPI) score is a well-established model for patients with aggressive non-Hodgkin lymphoma. The Follicular Lymphoma International Prognostic Index (FLIPI) is a similar model used for patients with follicular and other indolent lymphomas.
Several prognostic models have been developed to estimate prognosis in patients with non-Hodgkin lymphoma. The International Prognostic Index (IPI) score is a well-established model for patients with aggressive non-Hodgkin lymphoma. The Follicular Lymphoma International Prognostic Index (FLIPI) is a similar model used for patients with follicular and other indolent lymphomas.
![]() The clinical behavior and degree of aggressiveness can be used to categorize non-Hodgkin lymphoma into indolent and aggressive lymphomas. Patients with an indolent lymphoma usually have a relatively long survival, with or without aggressive chemotherapy. Although these lymphomas respond to a wide range of therapeutic approaches, few if any of these patients are cured of their disease. In contrast, aggressive lymphomas are rapidly growing tumors and patients have a short survival if appropriate therapy is not initiated. Most patients with aggressive lymphomas respond to intensive chemotherapy and many are cured of their disease.
The clinical behavior and degree of aggressiveness can be used to categorize non-Hodgkin lymphoma into indolent and aggressive lymphomas. Patients with an indolent lymphoma usually have a relatively long survival, with or without aggressive chemotherapy. Although these lymphomas respond to a wide range of therapeutic approaches, few if any of these patients are cured of their disease. In contrast, aggressive lymphomas are rapidly growing tumors and patients have a short survival if appropriate therapy is not initiated. Most patients with aggressive lymphomas respond to intensive chemotherapy and many are cured of their disease.
![]() Patients with localized follicular lymphoma can be cured with radiation therapy alone. Advanced follicular lymphoma is not curable, and there are many treatment options, including watchful waiting, extended-field radiation therapy, single-agent alkylating agents, anthracycline-containing combination chemotherapy, purine analogs, interferon-α, anti-CD20 monoclonal antibodies, and high-dose chemotherapy with hematopoietic stem cell transplantation.
Patients with localized follicular lymphoma can be cured with radiation therapy alone. Advanced follicular lymphoma is not curable, and there are many treatment options, including watchful waiting, extended-field radiation therapy, single-agent alkylating agents, anthracycline-containing combination chemotherapy, purine analogs, interferon-α, anti-CD20 monoclonal antibodies, and high-dose chemotherapy with hematopoietic stem cell transplantation.
![]() Patients with localized aggressive lymphomas can be cured with several cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine [Oncovin], prednisone) chemotherapy and involved-field irradiation. Patients with bulky stage II, stage III, or stage IV aggressive lymphomas can be cured of their disease with R-CHOP chemotherapy.
Patients with localized aggressive lymphomas can be cured with several cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine [Oncovin], prednisone) chemotherapy and involved-field irradiation. Patients with bulky stage II, stage III, or stage IV aggressive lymphomas can be cured of their disease with R-CHOP chemotherapy.
![]() Conventional-dose salvage therapy can induce responses in patients with aggressive lymphomas who relapse, but long-term survival and cure is uncommon. Some patients with aggressive lymphoma who relapse and respond to salvage therapy can be cured with high-dose chemotherapy and autologous hematopoietic stem cell transplantation.
Conventional-dose salvage therapy can induce responses in patients with aggressive lymphomas who relapse, but long-term survival and cure is uncommon. Some patients with aggressive lymphoma who relapse and respond to salvage therapy can be cured with high-dose chemotherapy and autologous hematopoietic stem cell transplantation.
Lymphomas are a heterogeneous group of malignancies that arise from malignant transformation of immune cells that reside predominantly in lymphoid tissues. They most commonly present as a solid tumor, but can sometimes present as circulating tumor cells in peripheral blood. The differing histology of lymphoma cells has led to classification of Hodgkin lymphoma (Reed–Sternberg cells) or non-Hodgkin lymphoma (B- or T-cell lymphocyte markers). Non-Hodgkin lymphomas (NHLs) are further classified into distinct clinical entities, which are defined by a combination of morphology, immunophenotype, genetic features, and clinical features. Chemotherapy is the mainstay of treatment in patients with lymphoma, especially those with widespread disease. Overall cure rates are high for many subtypes of lymphomas, even when patients present with advanced disease.
HODGKIN LYMPHOMA
Hodgkin lymphoma is a form of lymphoma, named after Thomas Hodgkin, who first described seven cases of a mysterious disease of the lymph system more than 150 years ago. Hodgkin lymphoma is fatal in more than 90% of the patients who are untreated for 2 to 3 years, and the cause is still unknown. The prognosis with treatment is generally good, but is not well predicted by stage alone. The International Prognostic Index (IPI) score was created to better predict an individual’s risk of recurrence, which in turn influences treatment decisions. Patients with Hodgkin lymphoma can be categorized into four prognostic groups: early favorable disease, early unfavorable disease, advanced favorable disease, and advanced unfavorable disease. These groups are defined by patient age, gender, tumor size and spread (tumor stage), presence or absence of systemic symptoms, and laboratory test results. When appropriate therapy is given, more than 75% of all newly diagnosed Hodgkin lymphoma patients can be cured. However, the success of treatment has not been without cost. The treatment programs are intense, technically demanding, and associated with considerable acute toxicity and long-term complications. The long-term effects, particularly secondary malignancies, account for a higher cumulative mortality than Hodgkin lymphoma 15 to 20 years after treatment. Long-term toxicities with standard chemotherapy regimens have been more fully documented in recent years and are shaping future therapies.1–3
Epidemiology and Etiology
Hodgkin lymphoma represents less than 1% of all known cancers in the United States. It is estimated that 9,290 new cases of Hodgkin lymphoma will be diagnosed in the United States in 2013, and there will be 1,180 deaths associated with Hodgkin lymphoma during this same period.4 This disease occurs slightly more frequently in males than in females. Once thought to be a disease of the young, it is now recognized that Hodgkin lymphoma exhibits bimodal distribution in industrialized countries. The first peak occurs in the third decade of life, with a small peak occurring after age 50.1,3 The 5-year overall survival for all stages of Hodgkin lymphoma is about 85%.5 Death rates as a consequence of recurrent Hodgkin lymphoma are less than those from other causes 15 years after treatment.6
The etiology of Hodgkin lymphoma is currently unknown, but laboratory and epidemiologic evidence support infectious exposure as a potential cause.7,8 Studies suggest an increased risk of Hodgkin lymphoma in patients who have been infected with the Epstein-Barr’s virus (EBV); and many patients experience EBV activation even before the onset of Hodgkin lymphoma. EBV is found in about 40% of all classical Hodgkin lymphoma cases, and it is frequently observed in cases of mixed cellularity and lymphocyte-depleted Hodgkin lymphoma.9 Reed–Sternberg cells (large, bilobate, multinuclear cells), the malignant cells in Hodgkin lymphoma, are linked to EBV. Immunosuppressed individuals are also at much higher risk to develop Hodgkin lymphoma. Such individuals include patients with congenital immunosuppression, solid-organ transplantation recipients, and human immunodeficiency virus (HIV)-infected patients. Although the risk of developing Hodgkin lymphoma is about sevenfold greater in patients with HIV, the level of CD4 may vary depending on the subtype of Hodgkin lymphoma.7
Genetic factors are also associated with an increased risk of Hodgkin lymphoma. The strongest evidence suggesting that genes are important in the etiology of Hodgkin lymphoma comes from identical twin studies, which show that the unaffected identical twin has almost a 100-fold increase in risk.10
Pathophysiology
Hodgkin lymphoma is a clonal malignant lymphoid disease of transformed lymphocytes. The malignant cell in Hodgkin lymphoma is known as the Reed–Sternberg cell named after Drs. Dorothy Reed and Carl Sternberg, who were credited with the first definitive microscopic description of Hodgkin lymphoma.1,11 Procedures to isolate and analyze Reed–Sternberg cells remain a challenge to scientists, due to the relatively small percentage (1% to 2%) of Reed–Sternberg cells that are found in the Hodgkin lymphoma mass.9 Fortunately, new laboratory techniques have led to significant progress in identifying the origin of the Reed–Sternberg cell. Single-cell polymerase chain reaction and DNA microarray analyses indicate that nearly all classic Hodgkin lymphoma cases and all nodular lymphocyte-predominant Hodgkin lymphomas have immunoglobulin gene rearrangements, which indicates a germinal center or postgerminal center B-cell origin.9,12 Interestingly, nearly all Reed–Sternberg cells fail to express B-cell specific cell surface proteins.
B-cell transcriptional processes are disrupted during malignant transformation, which prevents B-cell surface marker expression and production of immunoglobulin messenger ribonucleic acid. The normal cellular consequence of failure to express immunoglobulin is apoptosis, but because of alterations in the normal apoptotic pathways, cell survival and proliferation are favored. Reed–Sternberg cells overexpress nuclear factor-κ B, which is associated with cell proliferation and antiapoptotic signals. Infections with viral and bacterial pathogens upregulate nuclear factor-κ B and consequently are hypothesized to be involved with the etiology of Hodgkin lymphoma.1,9,12 This hypothesis is supported by the presence of EBV in many Hodgkin lymphoma tumors, but it is important to note that not all tumors are associated with EBV. Another signaling pathway, Janus kinase–signal transduction and transcription (JAK–STAT), has also been found to be active in Hodgkin lymphoma.1,9 As molecular techniques continue to improve, our understanding of the pathophysiology of Hodgkin lymphoma will also improve.
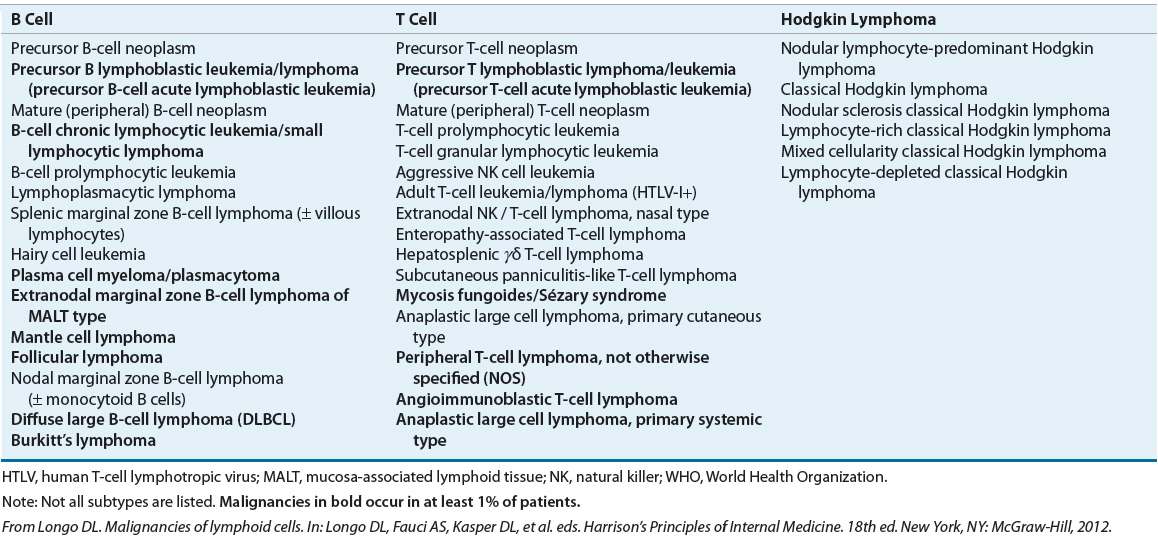
The histopathologic classification of Hodgkin lymphoma has undergone numerous changes over the past three decades. The current classification system is the 2008 World Health Organization (WHO) classification (see Table 109-1).13 This classification divides Hodgkin lymphoma into two major groups: classical Hodgkin lymphoma and nodular lymphocyte-predominant Hodgkin lymphoma, which constitute about 95% and 5% of cases, respectively. Classic Hodgkin lymphoma is further divided into four subtypes: nodular sclerosis, mixed cellularity, lymphocyte-depletion, and lymphocyte-rich. The subtypes in these classifications are based on characteristics of the Reed–Sternberg cell, the surrounding cells, and the connective tissue. Nodular sclerosis has features that make it distinct from the other three subtypes, which represent a continuum of background cellularity, with lymphocyte-predominance being the most cellular and lymphocyte-depletion being the least cellular. Nodular lymphocyte-predominant Hodgkin lymphoma is separated because of its distinct immunophenotype: CD15–, CD20+, CD30–, and CD45+ (the opposite of classical Hodgkin lymphoma). With the introduction of extensive staging, sophisticated radiotherapy, and effective combination chemotherapy, the prognostic value of these subtypes is becoming less clear. The true value of understanding these subtypes is likely tied to the pathogenesis of the disease and its potential prevention in the future.
TABLE 109-1 WHO Classification of the Mature B-Cell, T-Cell, and NK-Cell Neoplasms (2008)
Clinical Presentation
![]() Most patients with Hodgkin lymphoma present with a painless, rubbery, enlarged lymph node in the supradiaphragmatic area and commonly have mediastinal nodal involvement.14 Hodgkin lymphoma is occasionally diagnosed in an asymptomatic patient who has a mediastinal mass found with chest radiography or another imaging procedure. Asymptomatic adenopathy of the inguinal and axillary regions may be present at diagnosis but is less common (Fig. 109-1).1,14 Patients can also present with constitutional symptoms (B symptoms) before the discovery of lymph node enlargement, and these symptoms include fever, drenching night sweats, and weight loss. At diagnosis, these symptoms may appear in about 25% of all patients and up to 50% of patients with advanced disease. Patients may also experience other nonspecific symptoms including pruritus, fatigue, and development of pain after alcohol consumption at sites where nodes are involved.3 Extranodal manifestations, such as bowel and hepatic involvements, are much less common in Hodgkin lymphoma than NHL.1
Most patients with Hodgkin lymphoma present with a painless, rubbery, enlarged lymph node in the supradiaphragmatic area and commonly have mediastinal nodal involvement.14 Hodgkin lymphoma is occasionally diagnosed in an asymptomatic patient who has a mediastinal mass found with chest radiography or another imaging procedure. Asymptomatic adenopathy of the inguinal and axillary regions may be present at diagnosis but is less common (Fig. 109-1).1,14 Patients can also present with constitutional symptoms (B symptoms) before the discovery of lymph node enlargement, and these symptoms include fever, drenching night sweats, and weight loss. At diagnosis, these symptoms may appear in about 25% of all patients and up to 50% of patients with advanced disease. Patients may also experience other nonspecific symptoms including pruritus, fatigue, and development of pain after alcohol consumption at sites where nodes are involved.3 Extranodal manifestations, such as bowel and hepatic involvements, are much less common in Hodgkin lymphoma than NHL.1
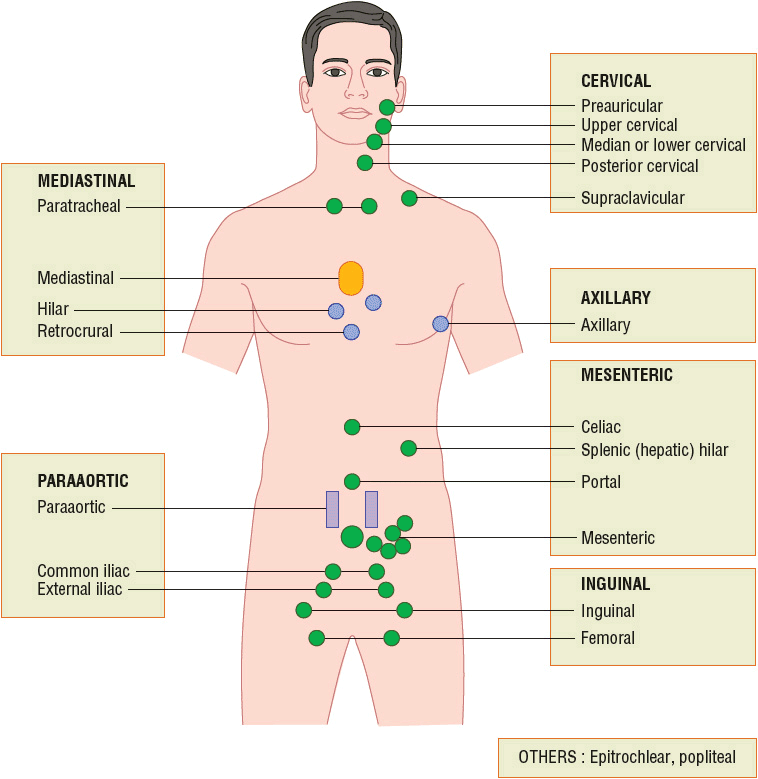
FIGURE 109-1 Areas of lymph nodes used in the staging of Hodgkin and non-Hodgkin lymphoma. Each rectangle corresponds to a nodal area.
Diagnosis, Staging, and Prognostic Factors
Diagnostic and staging procedures are based on recommendations made at the Ann Arbor and Cotswolds conferences and new scientific advances, as described in the National Comprehensive Cancer Network (NCCN) guidelines.15 The diagnosis and pathologic classification of Hodgkin lymphoma can only be made by review of a biopsy (preferably an excisional biopsy) of the enlarged node by an expert hematopathologist.
In addition to a careful physical examination and routine laboratory tests, chest radiography and computed tomography (CT) scans of the chest, abdomen, and pelvis are routinely performed. Furthermore, positron emission tomography (PET) plays an important role in the initial staging of Hodgkin lymphoma, as it has shown high sensitivity and specificity in the staging of the disease.16 The use of integrated PET-CT has further improved the staging of Hodgkin lymphoma given that it can provide more sensitive and specific imaging as compared with each imaging alone. The NCCN guideline recommends either an integrated PET-CT scan (preferred) or a PET scan with diagnostic CT for initial staging.15 Bone marrow biopsy is also recommended in patients with advanced-stage disease.
Staging can be based on clinical or pathologic findings. The clinical stage is based on all noninvasive procedures (history, physical examination, laboratory tests, and radiologic findings), whereas the pathologic stage is based on the biopsy findings of strategic sites (muscle, bone, skin, spleen, and abdominal nodes) with an invasive procedure such as a laparoscopy or laparotomy. Patients with extranodal disease (muscle, skin, bone, or Waldeyer ring) contiguous to involved nodes are classified with the subscript “E” in the Cotswolds staging system.15 As a result of improved imaging techniques, pathologic workup and staging that can be associated with toxicity is rarely performed.
CLINICAL PRESENTATION Hodgkin Lymphoma
The Ann Arbor staging classification, which was developed at the 1970 Ann Arbor conference, has proven to be a good workable scheme. At the Cotswolds meeting in 1989, the Ann Arbor classification was modified to account for new diagnostic techniques (e.g., CT and magnetic resonance imaging), and the understanding that prognosis is associated with the bulk of the disease and the number of involved nodal sites (see Table 109-2).3 After careful staging, about one-half of patients have localized disease (stages I, II, and IIE) and the remainder have advanced disease (stage III or IV). About 10% to 15% present with metastatic disease (stage IV). It is important to note that Hodgkin lymphoma appears to follow a predictable pattern of nodal spread that is not seen with the NHLs.14,15
TABLE 109-2 The Ann Arbor Staging Classification of Hodgkin Lymphoma

Patient prognosis is predominately driven by age and tumor stage. Patients older than ages 65 to 70 have a lower cure rate than younger patients. The difference in cure rates may be related to the frequent presence of comorbid diseases and decreased organ function in older patients, which impairs their ability to tolerate intensive chemotherapy.17 Stage is the other dominant factor in predicting survival; patients with limited-stage disease (stages I to II) have a 90% to 95% cure rate, while those with advanced disease (stages III to IV) have only a 60% to 80% cure rate.3,15
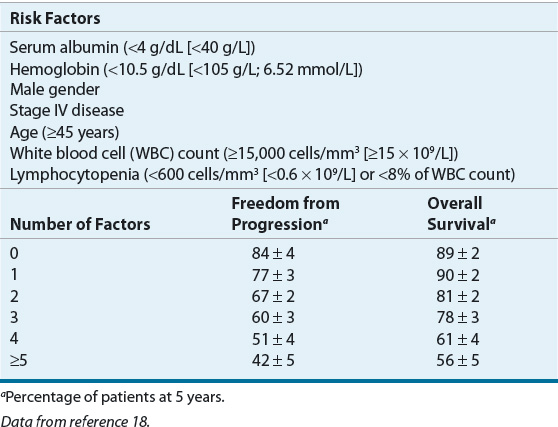
Seven adverse prognostic factors with similar impact on survival (each factor reduced survival by 7% to 8% per year) have been identified through an international collaborative effort. These factors can be combined to generate an International Prognostic Score (IPS) that can be used to predict progression-free and overall survival (see Table 109-3).18
TABLE 109-3 The International Prognostic Factors Project Score for Advanced Hodgkin Lymphoma
TREATMENT
Hodgkin Lymphoma
Desired Outcomes
The current goal in the treatment of Hodgkin lymphoma is to maximize curability while minimizing short- and long-term treatment-related complications. According to the Surveillance, Epidemiology, and End Results (SEER) database, the 5-year age-adjusted relative survival is greater than 80%.5 Therefore, the treatment goal of all stages of Hodgkin lymphoma should be cure.
General Approach to Treatment
Although multiple treatment modalities are used to treat Hodgkin lymphoma, surgery has a limited therapeutic role regardless of stage. It is, however, important for diagnosis (excisional biopsy), and on certain occasions, such as placement of a central line.
Combination chemotherapy is the primary treatment modality for most patients with Hodgkin lymphoma. In general, patients with early stage Hodgkin lymphoma are treated with combination chemotherapy and radiation, whereas patients with advanced-stage disease are treated with combination chemotherapy with or without radiation therapy. For patients with refractory or recurrent disease, salvage therapy consists of multiagent chemotherapy with or without high-dose chemotherapy and autologous hematopoietic stem cell transplantation (HSCT), which can be curative.1,3,19
Radiation is often an integral part of the treatment plan. Selected patients with early stage disease (usually nodular lymphocyte-predominant histology) can receive radiation as the only treatment modality, whereas most patients will receive chemotherapy and radiation. Although radiation is a local therapy, many patients with advanced disease will also receive radiation therapy to residual or bulky disease sites after chemotherapy. The major concern with radiation therapy is its long-term effects, such as cardiovascular disease and secondary malignancies, which commonly occur in the lung, breast, gastrointestinal tract, and connective tissue.20 To avoid these toxicities, several studies have been completed and others are ongoing to determine the optimal extent (radiation field) and dose of radiation.21,22 Radiation to a single field that contains Hodgkin lymphoma is called involved-field radiation; radiation to the involved field and a second uninvolved area is termed extended-field radiation or subtotal nodal irradiation; and radiation of all areas is called total nodal irradiation. When given with chemotherapy, involved-field radiation is usually used to avoid the increased toxicity associated with extended-field radiation.21 The following sections review treatment of early stage favorable disease, early stage unfavorable disease, advanced-stage favorable disease, advanced-stage unfavorable disease, and salvage therapy.
Treatment of Early Stage Favorable Disease
Patients with early stage favorable disease have stage IA or IIA disease and no adverse risk factors (extranodal disease, bulky disease, three or more sites of nodal involvement, or an erythrocyte sedimentation rate of ≥50 mm/h [≥13.9 μm/s]). In the past, extended-field radiation was considered to be the treatment of choice for stages IA and IIA disease. Although most patients were cured of their disease, it is associated with long-term toxicities due to large radiation fields, such as heart disease, pulmonary dysfunction, and secondary malignancies.3,20,23
In an effort to avoid the long-term effects of extended-field radiation and improve treatment results, several studies have evaluated a combined modality approach that involves the use of short-duration chemotherapy and involved-field radiation. Based on favorable results of these studies, most patients with early stage favorable disease are no longer treated with radiation alone.
Clinical trials comparing radiation alone to radiation plus chemotherapy show lower relapse rates in patients treated with combined modality therapy (radiation and chemotherapy), but no change in overall survival because of the availability of effective salvage therapy.22 Current trials focus on questions such as the optimal number of chemotherapy cycles, the volume of radiation that must be used to obtain optimal patient outcomes, and the role of PET scanning to individualize therapy. Emerging data also suggest that as few as two cycles of chemotherapy followed by involved-field radiation is sufficient in favorable, early stage disease patients. Different combination chemotherapy regimens have been used in these studies, and no one regimen is clearly superior to another.21–23
The current NCCN guidelines recommend that patients with early stage favorable disease be treated with two cycles of the Stanford V regimen (doxorubicin, vinblastine, mechlorethamine, etoposide, vincristine, bleomycin, and prednisone) or four cycles of the ABVD (doxorubicin [Adriamycin], bleomycin, vinblastine, and dacarbazine) regimen, followed by consolidative involved-field radiation.24 With this approach, 5-year progression-free and overall survival rates of >90% can be achieved.
Investigators are now focusing on novel strategies to minimize the long-term toxicities of Hodgkin lymphoma treatment, particularly among those patients who have good treatment prognosis. One recently published study compared the efficacy of two versus four cycles of ABVD and involved-field radiation in early stage favorable disease Hodgkin lymphoma patients. It was shown that treatment with two cycles was as effective and less toxic than four cycles of chemotherapy followed radiation therapy at 5 years.25 Such encouraging data would need to be confirmed with longer followup of the treatment.
Nodular lymphocyte-predominant Hodgkin lymphoma has been described as more indolent in nature, and better prognosis can be achieved when compared with classic Hodgkin lymphoma. The use of radiation alone for nodular lymphocyte-predominant Hodgkin lymphoma patients who choose to omit chemotherapy, or who cannot tolerate chemotherapy, does not appear to adversely affect survival.15 The disadvantage of radiation therapy alone as compared with combination chemotherapy and radiation is the higher relapse rate. Patients who relapse after radiation alone (20% to 25%) can be successfully salvaged with chemotherapy. If the decision is made to use radiation alone, extended-field radiation appears to be superior to involved-field radiation.26 It is shown that more extensive radiation reduces the risk of treatment failure at 10 years (31% vs. 43%), although it does not improve overall survival. However, the risk of long-term complications (such as secondary malignancies) is increased with the use of extended-field radiotherapy when compared with less extensive radiation fields.20
Treatment of Early Stage Unfavorable Disease
Patients with early stage disease who have certain features associated with a poor prognosis (B symptoms, extranodal disease, bulky disease, three or more sites of nodal involvement, or an erythrocyte sedimentation rate >50 mm/h [≥13.9 μm/s]) are defined as having unfavorable disease. Different groups or clinical trials have different definitions for unfavorable disease.22 Current guidelines recommend combined modality therapy (combination chemotherapy and involved-field radiation) to reduce the relapse rate and avoid the toxicity associated with extended-field radiation.15
![]() Although randomized trials show that combined modality therapy reduces the relapse rate in patients with early stage unfavorable disease, questions concerning the appropriate radiation volume, most effective chemotherapy regimen, and number of chemotherapy cycles remain.22 A number of studies have compared extended-field radiation to involved-field radiation. In one large trial conducted by the German Hodgkin Study Group (GHSG), patients with early stage unfavorable Hodgkin lymphoma treated with chemotherapy and involved-field radiation had similar freedom from treatment failure and overall survival as those treated with the same chemotherapy regimen and extended-field radiation.27 Because toxicity was greater with extended-field radiation, the accepted standard is chemotherapy and involved-field radiation.
Although randomized trials show that combined modality therapy reduces the relapse rate in patients with early stage unfavorable disease, questions concerning the appropriate radiation volume, most effective chemotherapy regimen, and number of chemotherapy cycles remain.22 A number of studies have compared extended-field radiation to involved-field radiation. In one large trial conducted by the German Hodgkin Study Group (GHSG), patients with early stage unfavorable Hodgkin lymphoma treated with chemotherapy and involved-field radiation had similar freedom from treatment failure and overall survival as those treated with the same chemotherapy regimen and extended-field radiation.27 Because toxicity was greater with extended-field radiation, the accepted standard is chemotherapy and involved-field radiation.
Different chemotherapy regimens and number of chemotherapy cycles were also compared in clinical trials. Mechlorethamine, vincristine, procarbazine, and prednisone (MOPP) was one of the first highly effective regimens introduced to treat Hodgkin lymphoma. MOPP or MOPP-like regimens were then given alternately or hybridized with a combination of ABVD. In advanced disease, ABVD was found to be less toxic than alternating MOPP/ABVD, and both were found to be superior to MOPP alone. Consequently, ABVD has now become the standard regimen used to treat patients with early stage unfavorable disease. It has established effectiveness in patients with advanced-stage disease and a favorable toxicity (both acute and chronic) profile.
Despite excellent results from treatment with ABVD and radiation, about 5% of patients do not respond to initial treatment and another 15% of patients will relapse following an initial response. Several studies have evaluated more aggressive regimens or more cycles of therapy. However, none of these regimens has proven to be more effective than ABVD, and each is associated with more toxicity. The current NCCN guideline lists the Stanford V regimen as an acceptable option, which also uses more drugs than the ABVD regimen.15
In summary, most patients with early stage disease will be treated with two to four cycles of ABVD chemotherapy and involved-field radiation. The number of cycles administered is based on the classification of favorable versus unfavorable disease. For patients who have unfavorable disease with bulky disease, ABVD followed by involved-field radiation. If patients attain complete remission after four cycles by restaging, they can be treated with involved-field radiation alone or two additional cycles of ABVD (total of six) followed by involved-field radiation. For patients who have unfavorable yet nonbulky disease, observation without involved-field radiation can be a treatment option after six cycles of ABVD.15 Current trials are evaluating the use of PET scans as biomarkers to individualize therapy and minimize the amount of therapy necessary for cure.16
Treatment of Advanced-Stage Disease
Advanced-stage disease consists of stages III and IV disease. In some studies, stage IIB with a large mediastinal mass or extranodal disease is also considered advanced-stage disease (Table 109-2). By definition, patients with stages III and IV disease have tumors on both sides of the diaphragm, which almost always precludes the use of radiation alone as a therapeutic modality. Intensive combination chemotherapy is the mainstay of treatment, although some patients will benefit from radiation following chemotherapy. The prognosis of advanced-stage disease is excellent with 5-year overall survival rates ranging from less than 56% to 90%. Prognostic factors have been identified and standardized to provide a more accurate individual prognosis (Table 109-3).18
Patients with advanced-stage Hodgkin lymphoma can be classified into two groups based on the number of prognostic factors present from the IPI (Table 109-3). Advanced-stage patients with three or fewer poor prognostic factors are considered to have favorable disease and an about 60% likelihood of being failure-free at 5 years with traditional combination chemotherapy. Advanced-stage patients with four or more poor prognostic factors are considered to have unfavorable disease and a less than 50% likelihood of being failure-free at 5 years with traditional combination chemotherapy. Because of the high treatment failure rate, the therapeutic goal for this group of high-risk patients is to improve antitumor control.
Combination Chemotherapy
One of the initial combination chemotherapy regimens introduced in the early 1960s that was shown to cure advanced Hodgkin lymphoma was the MOPP regimen (Table 109-4). MOPP chemotherapy was a mainstay of treatment for patients with stages III and IV advanced Hodgkin lymphoma.
TABLE 109-4 Combination Chemotherapy Regimens for Hodgkin Lymphoma
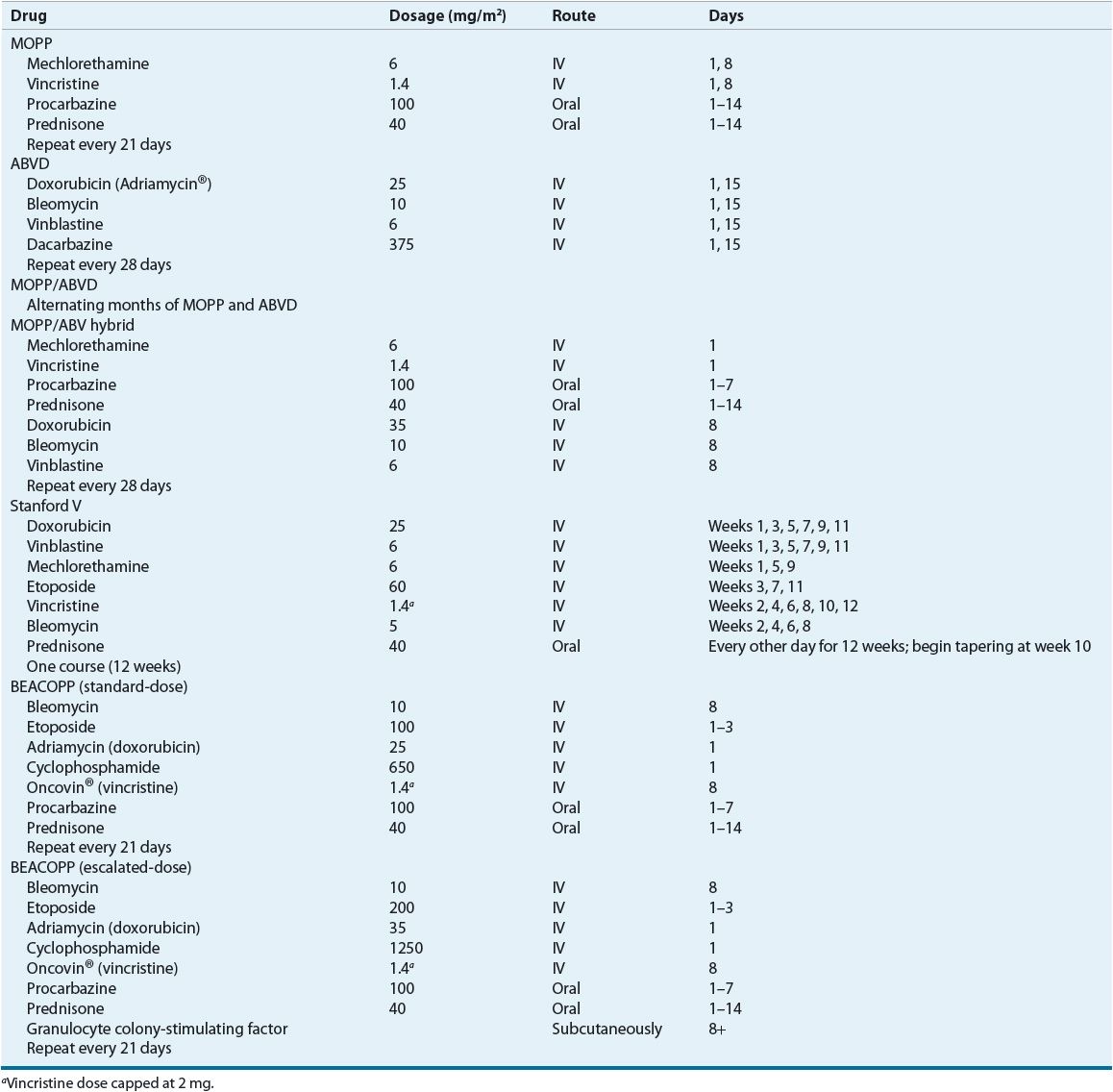
![]() The development of ABVD by Bonnadonna et al. at the Milan Cancer Institute about a decade later represents the next important step in the evolution of therapy for Hodgkin lymphoma (Table 109-4).28 ABVD was initially shown to be effective in treating MOPP failures and was later compared directly to MOPP in advanced disease, where it produced an 82% complete response rate, as compared to a 67% complete response rate with MOPP. Improved failure-free survival was demonstrated with ABVD, but no significant differences in 5-year overall survival were noted.29 Because ABVD was less toxic and provided similar or better outcomes than MOPP, it eventually replaced MOPP as the standard regimen for advanced-stage Hodgkin lymphoma.
The development of ABVD by Bonnadonna et al. at the Milan Cancer Institute about a decade later represents the next important step in the evolution of therapy for Hodgkin lymphoma (Table 109-4).28 ABVD was initially shown to be effective in treating MOPP failures and was later compared directly to MOPP in advanced disease, where it produced an 82% complete response rate, as compared to a 67% complete response rate with MOPP. Improved failure-free survival was demonstrated with ABVD, but no significant differences in 5-year overall survival were noted.29 Because ABVD was less toxic and provided similar or better outcomes than MOPP, it eventually replaced MOPP as the standard regimen for advanced-stage Hodgkin lymphoma.
In the early 1980s, the Goldie–Coldman hypothesis proposed that chemotherapy resistance was related to spontaneous mutation rates and the development of resistant clones. To test that hypothesis, researchers designed several clinical trials to evaluate the efficacy of alternating non–cross-resistant drug combinations in patients with Hodgkin lymphoma.30 The initial approach adopted by investigators was to alternate or combine the MOPP and ABVD regimens. When MOPP and ABVD (or doxorubicin [Adriamycin], bleomycin, vinblastine [ABV]) are combined in a monthly cycle, it is referred to as a hybrid regimen. Besides a potential benefit in efficacy, another potential benefit of alternating or hybrid regimens is the decreased risk of long-term toxicities. In the alternating MOPP/ABVD regimen, the cumulative doses of procarbazine and mechlorethamine are reduced by 50%, and the cumulative doxorubicin dose is reduced by 50%. In the hybrid regimen, the cumulative doxorubicin dose is reduced by 33%, and the cumulative bleomycin dose is reduced by 50%.
Several clinical trials have been performed to evaluate the efficacy of alternating or hybrid MOPP/ABVD regimens. The results of these trials show that alternating and hybrid regimens are superior to MOPP but not to ABVD.30 Another approach evaluated by researchers was the administration of sequential cycles of MOPP and ABVD (MOPP → ABVD). Results of an intergroup trial showed sequential MOPP and ABVD to be inferior to the MOPP/ABV hybrid regimen in terms of response and survival.31 In another randomized comparison trial of the MOPP/ABV hybrid regimen and ABVD, the complete remission rate, failure-free survival, and overall survival were similar between the two regimens.32 The latter trial was closed prematurely because of an increased number of treatment-related deaths and secondary malignancies in the patients who received the MOPP/ABV hybrid regimen.
More aggressive regimens such as Stanford V and bleomycin, etoposide, doxorubicin (Adriamycin), cyclophosphamide, vincristine (Oncovin), procarbazine, and prednisone (BEACOPP) have been evaluated as alternatives to MOPP or ABVD.19 The Stanford V regimen generated considerable interest because of the results of phase II trials.23,33 Stanford V, ABVD, and a MOPP/ABV hybrid-like regimen (mechlorethamine, vincristine, procarbazine, prednisone, epidoxorubicin, bleomycin, vinblastine, lomustine, doxorubicin, and vindesine [MOPPEBVCAD]) were compared in a randomized trial to determine the best regimen to support a reduced radiotherapy program.34 Five-year failure-free and progression-free survival were significantly worse for the Stanford V regimen as compared to the other two regimens. However, no significant differences in projected 5-year progression-free and overall survival were observed between Stanford V and ABVD in a recently published randomized trial of patients with advanced Hodgkin lymphoma.35 The investigators speculated that differences in the application of radiotherapy may explain the divergent results in the two randomized trials. More pulmonary toxicity occurred in the ABVD group, but other toxicities occurred more frequently in the Stanford V group.
The GHSG developed the BEACOPP regimens based on principles of dose density, dose intensity, and mathematical modeling. BEACOPP uses similar drugs as in the cyclophosphamide, vincristine, procarbazine, and prednisone (COPP)/ABVD regimen, but rearranges the drugs in a shorter 3-week cycle. Several different versions of BEACOPP have been developed: standard-dose BEACOPP, escalated-dose BEACOPP, and dose-dense BEACOPP (BEACOPP-14).36 Granulocyte colony-stimulating factor support is required for the escalated-dose BEACOPP and BEACOPP-14 regimens.
Several randomized trials have compared BEACOPP to other regimens.3,19 The GHSG conducted a randomized comparison of eight cycles of COPP/ABVD (alternating), BEACOPP, or an escalated-dose BEACOPP regimen in 1,201 patients with advanced Hodgkin lymphoma.37 Most patients had advanced favorable disease, and all patients received radiation to sites of bulky or residual disease after chemotherapy. Escalated-dose BEACOPP appears to be the most active regimen in this study with 10-year freedom from treatment failure at 82% and overall survival at 86%, but this regimen also appeared to be more toxic.38 Despite filgrastim support, 90% of patients in the escalated-dose BEACOPP group had grade IV leukopenia, as compared with 19% in patients in the COPP/ABVD arm and 37% in the standard-dose BEACOPP arm. The higher rate of acute toxicity did not translate into a higher risk of treatment-related fatalities (<2% for all three regimens). The risk of toxic deaths due to sepsis or acute cardiac events was higher in elderly patients receiving BEACOPP.39 Furthermore, the higher rate of leukemia in patients receiving escalated-dose BEACOPP (2.5% at 5 years followup, and 3% at 10 years followup) as compared with those in the COPP/ABVD arm (0.4%) are of concern. In another study, 370 patients with advanced Hodgkin lymphoma were randomized to receive six cycles of ABVD, six cycles of CEC (cyclophosphamide, lomustine, vindesine, melphalan, prednisone, epirubicin, vincristine, procarbazine, vinblastine, bleomycin), or four cycles of escalated-dose BEACOPP with two cycles of standard-dose BEACOPP.40 BEACOPP was superior to ABVD for 5-year failure-free survival (78% vs. 65%, P = 0.036) and progression-free survival (81% vs. 68%, P = 0.038), but 5-year overall survival was not significantly different between ABVD and BEACOPP. It appears that BEACOPP may be superior to ABVD in patients with high-risk advanced Hodgkin lymphoma (IPS ≥3). Higher rates of neutropenia and severe infections were observed with BEACOPP as compared with ABVD.
Finally, GHSG has conducted several trials to evaluate the optimal number and intensity of BEACOPP. In the HD12 study, eight cycles of escalated-dose BEACOPP were compared to four cycles of escalated-dose and four cycles of standard-dose BEACOPP (4 + 4 BEACOPP regimen) with or without radiation therapy. Freedom-from-treatment failure, progression-free survival, and overall survival were not different between the two treatment arms.41 Although the 4 + 4 BEACOPP regimen resulted in less acute hematologic toxicities during cycles five to six, this difference did not translate into a lower risk of treatment-related mortality. In the recently published HD15 noninferiority trial, eight cycles of escalated-dose BEACOPP was compared with six cycles of escalated-dose BEACOPP and eight cycles of BEACOPP-14.42 Freedom-from-treatment failure and overall survival were significantly better in the patients treated with six cycles of escalated-dose BEACOPP as compared with those treated with eight cycles of escalated-dose BEACOPP. No significant differences were observed between the eight cycles of escalated-dose BEACOPP versus BEACOPP-14. The risk of treatment-related mortality and secondary malignancies was also lower in patients treated with six cycles of escalated-dose BEACOPP. Of interest was the use of PET-guided radiotherapy in all of the treatment arms.
Clinical Controversy…
The results of these studies suggest that escalated-dose BEACOPP is superior to ABVD in the treatment of advanced Hodgkin lymphoma, but at the cost of more treatment-related toxicity. Some experts recommend that these more intensive regimens should only be considered in patients with high-risk disease because of the potentially higher risk of secondary malignancies. The NCCN guideline suggests that patients attaining a complete remission after four cycles of escalated-dose BEACOPP can be followed up with standard-dose BEACOPP and radiation therapy.15
Risk-Adapted Therapy
Patients with advanced-stage Hodgkin lymphoma can be classified into two groups based on the IPS (Table 109-3). Advanced-stage patients with three or fewer poor prognostic factors are considered to have favorable disease and an about 60% likelihood of being failure-free at 5 years with traditional combination chemotherapy.18 Advanced-stage patients with four or more poor prognostic factors are considered to have unfavorable disease and a less than 50% likelihood of being failure-free at 5 years with traditional combination chemotherapy. Because of the high treatment failure rate, the therapeutic goal for high-risk patients is to improve antitumor control.
One recently published study has reported the feasibility of a risk-adapted treatment approach based on the IPS, with the goal of reducing cumulative doses of chemotherapy in patients with low-risk Hodgkin lymphoma.43 Low-risk patients with early unfavorable disease and standard-risk patients with IPS of <3 were treated with two cycles of standard-dose BEACOPP, and high-risk patients with an IPS ≥3 were treated with two cycles of escalated-dose BEACOPP. After an interim gallium or PET/CT scan, patients with positive disease were given escalated-dose BEACOPP, while patients who had negative disease were given standard-dose BEACOPP. For all patients, the complete remission rate, 5-year event-free survival, and overall survival were 97%, 85%, and 90%, respectively. Although this was not a randomized study, the results support the use of a risk-adapted treatment modality in the treatment of advanced-stage Hodgkin lymphoma.
In summary, the NCCN guidelines suggest that ABVD or Stanford V should be considered for primary treatment for patients with advanced disease. Escalated-dose BEACOPP should be considered for patients with unfavorable disease because of increased efficacy. It is recommended that patients in advanced-stage disease with an IPS <4 should be treated with ABVD because of less acute toxicity, the absence of sterility, and a low risk of secondary acute myeloid leukemia/myelodysplastic syndrome.15
Radiation
The role of low-dose consolidative radiation when added to chemotherapy for the treatment of advanced-stage Hodgkin lymphoma is controversial.19,23 The rationale for its use is based on the radiosensitivity of Hodgkin lymphoma, a 20% to 40% relapse rate, and the tendency of Hodgkin lymphoma to relapse at sites of initial involvement.44 Many clinical trials have been conducted to evaluate the benefit of additional radiation in patients who have a complete response to combination chemotherapy. The results of these studies are inconsistent, and a meta-analysis of 14 randomized trials showed a modest improvement in disease control at 10 years, but no difference in overall survival.45 In one study, patients with advanced disease were randomized to receive either involved-field radiation after MOPP/ABV hybrid chemotherapy or no further therapy. Eight-year event-free survival reported for patients achieving a complete response randomized to receive radiation, no radiation, and a group of partial responders who received radiation were 73%, 77% and 76%, respectively. These results suggest that radiation provides no benefit for patients who achieve a complete remission with chemotherapy. Furthermore, radiation was also associated with a higher risk of secondary cancers (12.9% vs. 5.6% in the radiation and no radiation arms, respectively). It does, however, show a significant role for consolidative radiation in patients who have a partial response after chemotherapy.46
Summary
In summary, the standard treatment of advanced-stage favorable Hodgkin lymphoma is six to eight cycles of ABVD chemotherapy. Escalated-dose BEACOPP should be considered for patients with unfavorable disease. This risk-adapted approach should result in 70% to >90% of patients achieving a complete remission and 60% to 80% of patients being cured of their disease. No further treatment is needed for patients attaining a complete remission. Patients achieving a partial remission should receive consolidative radiation to residual sites of disease.
Treatment of Refractory or Relapsed Disease
![]() The goal of salvage therapy is cure regardless of the site(s) of recurrence or primary therapy. With the increasing use of chemotherapy with or without radiation, regardless of disease extent, the rate of primary refractory disease is decreasing. Patients who do not achieve a complete remission with the initial regimen are considered to have primary refractory disease. These patients have a poor prognosis when treated with salvage chemotherapy, and therefore should be offered autologous HSCT as a treatment option.47,48
The goal of salvage therapy is cure regardless of the site(s) of recurrence or primary therapy. With the increasing use of chemotherapy with or without radiation, regardless of disease extent, the rate of primary refractory disease is decreasing. Patients who do not achieve a complete remission with the initial regimen are considered to have primary refractory disease. These patients have a poor prognosis when treated with salvage chemotherapy, and therefore should be offered autologous HSCT as a treatment option.47,48
Patients who relapse after an initial complete response can be treated with the same regimen, a different potentially non–cross-resistant regimen, radiation, or high-dose chemotherapy and autologous HSCT (often preceded by conventional-dose chemotherapy). There are no data available from randomized trials comparing the cytoreductive regimens that are used before transplantation. However, as most patients are now treated with ABVD, doxorubicin should be avoided in salvage chemotherapy regimens if the cumulative dose has reached >400 mg/m2, particularly in those patients who have received mediastinal radiotherapy because they are at much higher risk of cardiotoxicity.
The response to salvage therapy depends on the extent and site of recurrence, previous therapy, and duration of initial remission. Patients who relapse after radiation therapy alone have a good chance of being cured with combination chemotherapy, although fewer patients are being treated with radiation alone. High response rates (60% to 87%) have been observed with salvage chemotherapy regimens.49 Other patient groups who have a favorable prognosis following salvage therapy include patients who experience a local recurrence in a nonirradiated location and those who relapse more than 1 year after completion of their initial chemotherapy. Patients who experience late relapses can be cured with retreatment with the same chemotherapy regimen, treatment with a different, potentially non–cross-resistant regimen, or high-dose chemotherapy and autologous HSCT.
Patients who have an early relapse (<1 year after treatment) generally respond poorly to standard-dose salvage chemotherapy. High-dose chemotherapy and autologous HSCT is more effective, but also produces a higher risk of treatment-related mortality. Therefore, the choice of salvage treatment should consider the patient’s tolerance for a particular set of chemotherapeutic agents and treatment approach (standard-dose chemotherapy vs. high-dose chemotherapy and autologous HSCT).47
High-dose therapy should be considered in patients who relapse within 12 months of initial remission and in those who are refractory to first-line chemotherapy.47 Although no single preparative regimen has been shown to be superior to another, most regimens do not include total-body irradiation because of its potential pulmonary toxicity. Most patients are already at higher risk for pulmonary toxicity because of previous exposure to one or more of the following: bleomycin, thoracic radiation, and nitrosoureas. Long-term effects of high-dose chemotherapy and autologous HSCT were elucidated in a recent study. Among 218 Hodgkin lymphoma patients who were treated with high-dose chemotherapy and autologous HSCT, 70% survived over 2 years. A total of 15 patients were diagnosed with a second malignancy and the median time from autologous HSCT to secondary malignancy diagnosis was 9 years. The risk of secondary malignancy is possibly caused by high-dose etoposide in the rescue regimen, as well as those patients who were initially treated with MOPP chemotherapy.50
Brentuximab vedotin is an antibody-drug conjugate (ADC) comprising an anti-CD30 antibody conjugated by a protease cleavable linker to a potent antimicrotubule agent, monomethyl auristatin E (MMAE). After binding of the ADC to CD30 on the cell surface, the ADC-CD30 complex is internalized. This leads to the release of MMAE via proteolytic cleavage in the lysosomal compartment. Tubulin binding by MMAE disrupts the microtubule network, which can lead to apoptotic death of the cancer cells.51 In a pivotal multicenter phase II study of 102 patients with relapsed or refractory Hodgkin lymphoma after HSCT, objective responses and complete remissions was observed in 75% and 34% patients, after a median followup of 9 months. Common toxicities associated with brentuximab vedotin include neuropathy, neutropenia, nausea, and fatigue.52 Based on these results, the FDA recently approved brentuximab vedotin (Adcetris®) for the treatment of patients with Hodgkin lymphoma after failure of HSCT or in patients who have failed at least two prior chemotherapy regimens and are not candidates for HSCT.
Long-Term Complications
A variety of acute and chronic toxicities may occur as a result of treatment for Hodgkin lymphoma. Long-term complications of radiation therapy, chemotherapy, and combined modality therapy have become more evident as the curability and long-term survival of Hodgkin lymphoma patients has improved.1–3,20 Gonadal dysfunction (including sterility and hypothyroidism), secondary malignancies, and cardiopulmonary diseases have become important considerations in the treatment of this malignancy. Almost all men and up to 50% of premenopausal women treated with six cycles of regimens containing alkylating agents become sterile. This appears to be a dose-related phenomenon. For men, even a single dose of nitrogen mustard or chlorambucil can cause sterility, so if fertility is a major concern, ABVD may be the best alternative.50
The risk of secondary malignancies is increased about threefold in long-term survivors of Hodgkin lymphoma. The risk of developing leukemia carries the highest increase in risk and is seen with radiotherapy, chemotherapy, and chemoradiotherapy. Solid tumors, including breast cancers, gastrointestinal cancers, and lung cancers are also likely to develop more than 10 years after the completion of treatment.53–55 A recently published British cohort study suggested that unlike radiotherapy, which may increase the occurrence of cancer at almost all anatomic sites, chemotherapy is associated with an increased risk of leukemia, NHL, and lung cancer.55 However, studies that evaluate the risk of secondary malignancies (and other complications) must be interpreted cautiously because many factors probably contribute to the development of secondary malignancies.56 In addition, much of the long-term complication data are derived from patients who were treated with older regimens and extensive field radiotherapy, which are no longer commonly used in clinical practice. Furthermore, as minimal data are currently available on the appropriate followup duration and procedures to monitor for long-term effects, many of the recommendations in the NCCN guideline are based on clinical practice. Monitoring and followup should be personalized and patient-specific, after assessing a patient’s risks for long-term complications.15
NON-HODGKIN LYMPHOMA
The NHLs are a heterogeneous group of lymphoproliferative disorders that affect individuals from early childhood to late adulthood. Advances in molecular biology techniques and our understanding of the human immune system have led to major progress in understanding the pathogenesis and treatment of the lymphomas. NHLs are classified into distinct clinical entities that are defined by a combination of morphology, immunophenotype, genetic features, and clinical features. These differences influence the natural history, and approach and response to treatment. The use of extensive combination chemotherapeutic regimens shows dramatic improvement in survival and cure in patients with a disease that was once considered incurable. The 5-year survival rate for patients with NHL has increased from 48% to 71% over the past 25 years, and the mortality rate actually declined from 1997 to 2004.4,5 Further improvement in survival is anticipated with the continued expansion of our therapeutic armamentarium, including high-dose chemotherapy and biologic therapy.
Epidemiology and Etiology
NHL is the fifth most common cause of newly diagnosed cancer in the United States and accounts for about 4% of all cancers. An estimated 69,740 new cases will be diagnosed in 2013, and it is estimated that 19,020 people will die from NHL during this same period.4 Although the average age of patients at the time of diagnosis is about 67 years, NHL can occur at any age. The incidence rate generally increases with age, and is higher in men than in women and in whites than in blacks.5 The age-adjusted incidence rate of NHL increased by more than 80% in the United States since the early 1970s, from about 11 cases per 100,000 in 1975 to about 20 cases per 100,000 in 2003 and 2004.5 The incidence of NHL increased by 3% to 4% from 1975 to 1991, but appears to have stabilized since reaching its peak in 1994. The increased incidence of NHL over the past three decades is second only to melanoma and has been referred to as an epidemic of NHL. Although the increase has been noted particularly among the elderly and patients with acquired immune deficiency syndrome (AIDS), much of it cannot be explained by known risk factors.
The etiology of NHL is unknown, although several genetic diseases, environmental agents, and infectious agents are associated with the development of NHL.7,57 An increased incidence of NHL is seen in many congenital and acquired immunodeficiency states, supporting the role of immune dysregulation in the etiology of NHL.57 Patients with congenital immunodeficiency disorders such as Wiskott-Aldrich’s syndrome and ataxia telangiectasia, acquired immunodeficiency disorders such as AIDS, and those receiving chronic pharmacologic immunosuppression in the setting of solid-organ transplantation are predisposed to the development of NHL. Autoimmune diseases (Hashimoto’s thyroiditis, Sjögren’s syndrome) cause chronic inflammation in the mucosa-associated lymphoid tissue (MALT), which predisposes patients to subsequent lymphoid malignancies. Other autoimmune diseases, such as systemic lupus erythematosus and rheumatoid arthritis, are also associated with the development of NHL, but the use of immunosuppressive agents in these diseases makes the pathologic cause less clear.
Certain infections are associated with the development of lymphoma.7 EBV was discovered in cell lines from tumors of patients with African (endemic) Burkitt lymphoma, and EBV DNA is associated with nearly all cases of endemic Burkitt lymphoma. However, EBV is associated with sporadic Burkitt lymphoma in 15% to 85% of cases. EBV is also associated with posttransplant lymphoproliferative disorders and some lymphomas in patients with AIDS or congenital immunodeficiencies. The human T-cell lymphotropic virus type 1 was the first human retrovirus associated with a malignancy. Infection with human T-cell lymphotropic virus type 1, especially in early childhood, is strongly associated with an aggressive form of T-cell lymphoma, known as adult T-cell leukemia/lymphoma. Human T-cell lymphotropic virus type 1 is endemic in parts of southern Japan, Africa, South America, and the Caribbean. In endemic areas, more than 50% of all NHL cases are adult T-cell leukemia/lymphoma. A third virus associated with NHL is human herpes virus 8 (also referred as Kaposi sarcoma–associated herpesvirus [KSHV]). This virus was originally isolated from Kaposi sarcoma lesions in AIDS patients. Gastric infection with Helicobacter pylori, a gram-negative bacteria that leads to chronic gastritis, is associated with gastric MALT lymphomas. Finally, hepatitis C virus has been associated with splenic and nodal marginal zone lymphomas.
A number of physical agents are also associated with the development of NHL.57 Exposure to herbicides, particularly phenoxyl herbicides, is associated with the development of NHL. These observations may explain why certain occupations, such as farmers, forestry workers, and agricultural workers, are associated with a higher risk of NHL. Exposure to lawn-care pesticides is also increasing in the general population. A higher risk of NHL is also associated with exposure to other chemical solvents and dyes, exposure to radiation from nuclear explosions, and high intake of meats and dietary fats. Smoking or alcohol consumption is not strongly associated with an increased risk of NHL.
Molecular Abnormalities
Chromosomal translocations have become a hallmark of many lymphoid malignancies.58,59 The presence of these specific translocations can be helpful in the diagnosis and classification of lymphoid malignancies. The mechanisms leading to the translocations are unknown, but they usually involve the antigen receptor loci. In contrast to most myeloid and some lymphoid leukemias, NHLs usually place a structurally intact cellular protooncogene under the regulatory influence of highly expressed immunoglobulin or T-cell receptor genes, leading to effects on cell growth, cellular differentiation, or apoptosis. The most common chromosomal translocations involve t(8;14), t(14;18), and t(11;14); each translocation involves the immunoglobulin heavy-chain gene locus on chromosome 14 at 14q32. The translocation t(8;14) that involves c-MYC, a well-characterized oncogene clearly associated with malignancy, is implicated in nearly all cases of Burkitt lymphoma. The translocation t(14;18) that involves BCL-2, one of several putative B-cell lymphoma–associated oncogenes, is found in about 90% of cases of follicular B-cell lymphomas. The translocation t(11;14) that involves BCL-1 is found in about 70% of patients with mantle cell lymphoma. Another putative B-cell lymphoma–associated oncogene, BCL-6, is found in about one-third of diffuse large B-cell lymphomas.
Although mutations in the p53 tumor suppressor gene have been recognized in many human neoplasms, such mutations have not been consistently found in patients with lymphoma, which suggests that it may occur late in malignant evolution.
Because of their role in the pathogenesis of lymphoma, oncogenes are attractive molecular targets for the development of new and novel therapies.60
Pathology and Classification
NHLs are neoplasms derived from the monoclonal proliferation of malignant B or T lymphocytes and their precursors. About 85% to 90% of NHLs in the United States are of B-cell origin.57 Proliferation of malignant cells results in the replacement of the normal cells and architecture of lymph nodes or bone marrow with a relatively uniform population of lymphoid cells. The classification of NHLs has evolved over the past five decades, as advances in immunology and genetics have allowed scientists to recognize a number of previously unrecognized subtypes of NHLs (Table 109-5).61,62 The current classification schemes characterize the NHLs according to the cell of origin (B cell vs. T cell), clinical features, and morphologic features. Additional immunohistochemical markers, cytogenetic features, and genotypic characteristics may help to further classify NHL into subtypes.
TABLE 109-5 Evolution in the Classification of Non-Hodgkin Lymphomas

Morphology
The macroscopic and microscopic appearance of the involved tissue remains one of the most important factors in the diagnosis and classification of NHLs.61,62 In the 1950s, Rappaport et al. proposed a morphologic classification of malignant lymphomas based on two features: that the malignant cell would disrupt the nodal architecture in a nodular or diffuse manner, and that lymphomas of histiocytic origin existed. The Rappaport classification gained rapid acceptance in the United States because of its precision, simplicity, and prognostic significance. Application of the system divided NHLs into those with large (i.e., incorrectly called “histiocytes”) or small cells, with or without a nodular (i.e., follicular) growth pattern.
Immunology
In the 1970s, it became apparent that NHLs were tumors of the immune system and were derived from B or T lymphocytes. With the availability of techniques using antibodies to antigens on the surface of lymphoid cells (i.e., immunophenotype) and cytochemical assays, expert pathologists independently developed new classification schemes for NHL in the 1970s and 1980s.61,62 The Kiel classification was based primarily on the work of Lennert in Germany and became widely used in Europe. In North America, the Lukes and Collins classification scheme was used briefly, but was soon superseded by the Working Formulation. Like the Rappaport classification, divisions within the Working Formulation were based largely on cell size (large [histiocytic] vs. small [lymphocytic]), cell shape (round vs. not round), and growth pattern (follicular [nodular] vs. diffuse). Both the Kiel and Working Formulation classification schemes considered the histologic grade of the tumor, but only the Working Formulation considered actual survival curves of patients with the various subtypes of NHL. Low-grade indicated longer median survival (i.e., indolent) whereas intermediate-grade and high-grade indicated shorter median survival (i.e., aggressive). In the 1980s and early 1990s, the Working Formulation became the most widely used classification scheme in North America. It was based on the premise that NHL was a single disease with a range of histologic grades and clinical aggressiveness.
New Disease Entities
In the 1980s and early 1990s, rapid advances in immunology and genetics allowed scientists to recognize a number of previously unrecognized subtypes of NHLs. Cytogenetic and molecular genetic analyses identified the presence of many chromosomal translocations, oncogenes, and their gene products in patients with NHL (see Molecular Abnormalities later in this chapter). In addition, diseases that would have been lumped together as low-grade or intermediate/high grade in the Working Formulation showed marked differences in survival, which prompted scientists to reevaluate lymphoma classification schemes.
Information from these studies allowed scientists to further classify B-cell lymphomas as malignant expansions of cells from the germinal center, mantle zone, or marginal zone of normal lymph nodes.61,63 Germinal centers are complex structures that form in the spleen and lymph nodes in response to antigenic challenge. In addition to B cells, germinal centers contain antigen-presenting cells and helper T cells that cooperate in mediating the B-cell changes that result in a more potent secondary immune response. Malignant transformation often occurs or is initiated in germinal center B cells. Follicular, Burkitt, and most large cell lymphomas are believed to be tumors of germinal center B cells. Three histologically distinct microenvironments have been described within the germinal center: a mantle zone surrounding interior, dark, and light zones. The mantle zone contains small resting B cells that have not been exposed to antigens (naïve). Tumors of cells from the mantle zone are usually clinically indolent and histologically low grade. Antigen-triggered activation of the densely packed B cells of the dark zone causes cells to proliferate and subjects genomic DNA to somatic hypermutation. Surviving clones from within the dark zone then enter the light zone where proliferation slows and affinity selection occurs. During affinity selection, only cells with surface immunoglobulin receptors with high affinity for the antigen survive. Antigen-specific B cells generated in the germinal center reaction leave the follicle and reappear in the outer mantle zone, to form a marginal zone. Marginal zones are particularly prominent in mesenteric lymph nodes, Peyer’s patches, and the spleen. These postgerminal center B cells include memory B cells of the marginal zone and plasma cells. Marginal cell B-cell lymphomas tend to be indolent and may be either extranodal or nodal; extranodal marginal cell B-cell lymphomas are also referred to as MALT lymphomas.
T-cell lymphomas can be classified on the basis of antigen expression as either precursor (thymic) or mature (peripheral) in origin. These classifications clinically translate to precursor lymphoblastic lymphomas or to a heterogeneous group of peripheral T-cell lymphomas. Tumors of natural killer or natural killer-like T cells are uncommon.
The International Lymphoma Study Group, an informal group of 19 hematopathologists from the United States, Europe, and Asia, adopted a new approach to lymphoma classification in 1993. Because it represented a revision of current or prior European and American lymphoma classifications, it was called the Revised European-American Classification of Lymphoid Neoplasms (REAL). The REAL classification system is based on the principle that a classification is a list of “real” disease entities, which are defined by a combination of morphology, immunophenotype, genetic features, and clinical features.61,62 The relative importance of each of these criteria for both definition and diagnosis differs among different diseases. Morphology is always important, and some diseases are primarily defined by morphology alone (e.g., follicular lymphoma), although immunophenotype can be helpful in difficult cases. Some diseases have a specific immunophenotype (e.g., mantle cell lymphoma, small lymphocytic lymphoma) that is virtually diagnostic of that disease. A specific genetic abnormality is important in some lymphomas—t(11;14) in mantle cell lymphoma, t(8;14) in Burkitt lymphoma, and t(14;18) in follicular lymphoma—whereas other lymphomas lack specific genetic abnormalities (e.g., MALT lymphoma, diffuse large B-cell lymphoma). Finally, other lymphomas consider clinical features (e.g., extranodal vs. nodal presentation in marginal zone lymphoma and peripheral T-cell lymphoma).
Since 1995, members of the European and American Hematopathology societies have worked to develop a new WHO classification of hematologic malignancies. The final classification was published in 2001 and revised in 2008.13,61,62 The WHO classification uses an updated version of the REAL classification and expands the principles of the REAL classification to the classification of myeloid and lymphoid malignancies.
![]() The 2008 WHO classification categorizes lymphoid malignancies into two major categories: B-cell lymphomas and T-cell (and natural killer cell) lymphomas (Table 109-1).13,61,62 B-cell lymphomas represent about 85% to 90% of all NHLs. Lymphomas within each category can be divided into malignancies of precursor or mature cells. Hodgkin lymphoma and multiple myeloma are now recognized as mature B-cell neoplasms. The WHO classification uses the term grade to refer to histologic parameters such as cell and nuclear size, density of chromatin, and proliferation fraction, and the term aggressiveness to denote clinical behavior of a tumor. This classification scheme includes both lymphomas and lymphoid leukemias because there is no distinction between the solid and circulating forms of these diseases. The WHO classification includes several previously unrecognized types of lymphomas, and new entities not specifically recognized in the Working Formulation account for about 20% to 25% of the cases.
The 2008 WHO classification categorizes lymphoid malignancies into two major categories: B-cell lymphomas and T-cell (and natural killer cell) lymphomas (Table 109-1).13,61,62 B-cell lymphomas represent about 85% to 90% of all NHLs. Lymphomas within each category can be divided into malignancies of precursor or mature cells. Hodgkin lymphoma and multiple myeloma are now recognized as mature B-cell neoplasms. The WHO classification uses the term grade to refer to histologic parameters such as cell and nuclear size, density of chromatin, and proliferation fraction, and the term aggressiveness to denote clinical behavior of a tumor. This classification scheme includes both lymphomas and lymphoid leukemias because there is no distinction between the solid and circulating forms of these diseases. The WHO classification includes several previously unrecognized types of lymphomas, and new entities not specifically recognized in the Working Formulation account for about 20% to 25% of the cases.
The WHO classification has broad clinical implications. The WHO Clinical Advisory Committee has agreed that clinical groupings of lymphoid neoplasms into prognostic categories are neither necessary nor desirable because such arbitrary groupings are of no practical value and may be misleading.64
Clinical Presentation
![]() Patients with NHL present with a wide variety of symptoms, depending on the site of involvement and whether tumor involvement is nodal or extranodal. Sites of involvement and dissemination of the malignant cells can sometimes be predicted based on the cell of origin and the tendency of tumors to frequently disseminate to areas where the normal counterparts of the lymphoma cells are located. For example, B-cell lymphomas involve areas of the lymphoid system normally populated by B-lymphocytes, such as lymph nodes, spleen, and bone marrow. T-cell lymphomas commonly disseminate to various extranodal sites, such as the skin and lungs.59
Patients with NHL present with a wide variety of symptoms, depending on the site of involvement and whether tumor involvement is nodal or extranodal. Sites of involvement and dissemination of the malignant cells can sometimes be predicted based on the cell of origin and the tendency of tumors to frequently disseminate to areas where the normal counterparts of the lymphoma cells are located. For example, B-cell lymphomas involve areas of the lymphoid system normally populated by B-lymphocytes, such as lymph nodes, spleen, and bone marrow. T-cell lymphomas commonly disseminate to various extranodal sites, such as the skin and lungs.59
Most patients present with peripheral lymphadenopathy. The lymphadenopathy may be either localized or generalized, and the involved nodes are often painless, rubbery, and discrete, and usually located in the cervical and supraclavicular regions as in Hodgkin lymphoma (Fig. 109-1). Rapid and progressive lymphadenopathy is more characteristic of aggressive lymphomas. Waxing and waning of lymph nodes, including their complete disappearance and reappearance, is more characteristic of indolent lymphomas. Massive lymphadenopathy can sometimes lead to organ dysfunction. For example, patients with NHL may present with acute renal failure from retroperitoneal adenopathy causing ureteral obstruction or from metabolic abnormalities such as hyperuricemia with uric acid nephropathy.
About 40% of patients with NHL present with fever (temperature >38°C [100.4°F]), weight loss (unexplained weight loss of 10% of body weight over the past 6 months), or night sweats (drenching night sweats). If one or more of these symptoms is present, the patient is noted to have B symptoms, and a B is added to the stage of disease (discussed in the Diagnosis, Staging, and Prognostic Factors section under Hodgkin Lymphoma earlier in this chapter). B symptoms are more commonly observed in patients with aggressive NHLs.



