Lymphoblastic Leukemia/Lymphoma
In the World Health Organization (WHO) classification, the commonly used acute lymphoblastic leukemia (ALL) and lymphoblastic lymphoma (LBL) are combined into the term lymphoblastic leukemia/lymphoma. This reflects the fact that these terms refer to a biologically very similar neoplasm, and the distinction between ALL or LBL can be, in part, arbitrary (1,2). Acute lymphoblastic leukemia is used to designate a patient with extensive peripheral blood and bone marrow involvement. Lymphoblastic lymphoma is used to designate a patient with lymph node or extranodal site–based disease without leukemia (although occasional blasts in blood may be present) and minimal involvement (<25%) or absence of bone marrow disease (1,2).
Although combining ALL and LBL into one term is both sensible and convenient, it is reasonable to hypothesize that some differences may exist between lymphoblastic neoplasms that present as either ALL or LBL. These differences may be at the DNA level or at the level of gene expression, and the terms ALL and LBL may again be unlinked in future versions of lymphoma classifications.
In the French-American-British (FAB) classification, cases of ALL were subdivided into three categories L1, L2, and L3, based on their cytologic features (3). With the benefit of current knowledge, it is clear that the L1 and L2 categories do not correlate with clinical, immunophenotypic, or genetic findings. In contrast, almost all cases of L3 are of mature B-cell lineage and correspond to the leukemic phase of Burkitt lymphoma (see Chapter 69). As a result, neoplasms classified in the past as ALL of FAB-L3 type are not included in the category of lymphoblastic leukemia/lymphoma.
Both immunophenotype and genetic alterations are essential to the classification, prognosis, and treatment of patients with lymphoblastic leukemia/lymphoma. Therefore, all cases of ALL/LBL need to be immunophenotyped, and cytogenetic and/or molecular studies are an essential part of the diagnostic workup. Lymphoblastic neoplasms of T- or B-cell lineage are recognized as different diseases, and both the T- and B-cell categories, as currently classified, are markedly heterogeneous at the genetic level. Recent data also suggest that a subset of ALL/LBL is of natural killer (NK)-cell lineage.
Precursor T-Cell Lymphoblastic Leukemia/Lymphoma
Definition
Precursor T-cell (pre-T) lymphoblastic leukemia/lymphoma refers to a neoplasm of immature lymphoid cells (lymphoblasts) of T-cell lineage (1). Patients with ALL/LBL most often present with clinical findings typical of lymphoma.
Synonyms
T-cell acute lymphoblastic leukemia (FAB L1 or L2); convoluted T-cell lymphoma; lymphoma of precursor or immature T-lymphocytes.
Epidemiology and Pathogenesis
In the Surveillance, Epidemiology, and End Results (SEER) database for 1992–2001, lymphoblastic leukemia/lymphoma represented 5.3% of all lymphoid neoplasms in the United States (4). Unfortunately, approximately 40% of cases were added to the SEER database without immunophenotypic data. Assuming that the neoplasms with immunophenotypic data reflect the population as a whole, pre-B neoplasms outnumber pre-T neoplasms by a ratio of 3:1. Extrapolating the data to include all cases in the database, the age-adjusted incidence rate for pre-T ALL/LBL in the United States is approximately 0.40 per 100,000 person-years (4). This corresponds to
approximately 1,000 cases per year in the United States. In a Norwegian study for the period 1985–2004, restricted to patients 15 years of age or older, the incidence of pre-T ALL/LBL was 0.20 per 100,000 per year (5).
approximately 1,000 cases per year in the United States. In a Norwegian study for the period 1985–2004, restricted to patients 15 years of age or older, the incidence of pre-T ALL/LBL was 0.20 per 100,000 per year (5).
Pre-T ALL/LBL affects whites more often than African Americans or Asians, and males are more often affected. The male-to-female ratio in the SEER study was highest in whites, 2.4:1 (4). Pre-T ALL/LBL is most common in adolescents and young adults under the age of 40 years. In the study from Norway, the median patient age was 28 years (5). Rarely, infants can present with the clinical picture of pre-T LBL (6). However, most young children or elderly adults with pre-T ALL/LBL present with acute leukemia.
In children and adolescents, up to 85% of all patients with pre-T lymphoblastic leukemia/lymphoma present as LBL with lymphadenopathy, a mediastinal mass, or both without leukemia. In adults, approximately 75% of patients present as LBL. However, both children and adults commonly go on to develop acute leukemia at some time during their clinical course, often rapidly. In the elderly (over 60 years of age), patients uncommonly present with a mediastinal mass and more often present as pre-T ALL (7). Pre-T LBL represented 1.7% of all non-Hodgkin lymphomas in the Non-Hodgkin’s Lymphoma Classification Project (8).
Based predominantly on immunophenotypic analysis, it appears that normal T cells acquire antigens in sequence as they mature (or differentiate), and that patterns of antigen expression can be grouped into sequential stages (9). This normally occurs in the thymus. Furthermore, individual cases of pre-T ALL/LBL express antigens that correspond to stages of maturation. These findings suggest that cases of pre-T ALL/LBL arise from thymocytes and that the neoplastic cells are frozen at a certain stage of maturation. Presumably, this stage corresponds to the normal stage of thymocyte maturation at the time neoplastic transformation occurred.
The etiology and pathogenesis of pre-T ALL/LBL are unknown. However, it is clear that this disease is heterogeneous at the molecular level, representing more than one disease or disease subsets. For example, gene expression profiling of childhood pre-T ALL cases showed four major molecular types (10). Furthermore, these molecular types are associated with distinct stages of arrested T-cell maturation and are correlated with cytogenetic abnormalities.
A subset of patients with pre-T ALL/LBL appears to have an inherited predisposition to pre-T ALL/LBL. For example, patients with the hereditary disorder ataxia-telangiectasia have an increased risk of developing pre-T ALL (11).
Clinical Findings
Patients who present with the clinical picture of pre-T LBL tend to be adolescents and relatively young adults (1,8,12,13,14,15). These patients present with widespread lymphadenopathy that is usually preferentially located above the diaphragm. Lymph nodes in the neck, supraclavicular region, and axillae are most often involved. A mediastinal mass is common, present in 50% to 75% of patients, and can cause superior vena cava syndrome or cardiac tamponade (1,12,13,14,15). Over 90% of patients have Ann Arbor clinical stage III or IV disease. Hepatomegaly and splenomegaly are often present, although less commonly than in patients who present with leukemia. The disease can involve a wide number of extranodal sites, either at time of diagnosis or during the clinical course. The so-called sanctuary sites—the central nervous system and gonads (Fig. 59.1)—can be involved, as well as a wide variety of extranodal sites. However, the gastrointestinal tract and intestinal lymph nodes are involved relatively infrequently. Peripheral blood involvement by lymphoblasts is observed in up to one-third of patients with pre-T LBL, and conversion to full-blown leukemia is common during the clinical course (1,13,14,15). In general, the white blood cell count is lower in patients presenting as pre-T LBL compared with patients presenting as pre-T ALL (1,14).
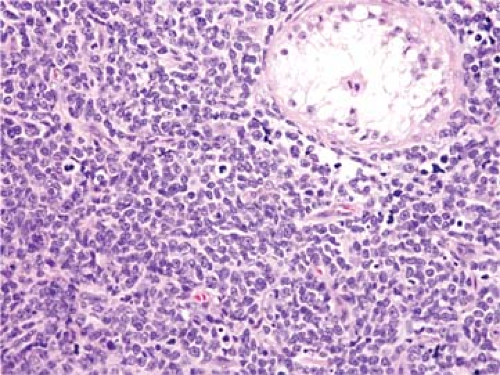 Figure 59.1. Precursor T-cell lymphoblastic leukemia/lymphoma involving testis. Hematoxylin-eosin stain. |
Patients who present with the clinical picture of pre-T ALL may complain of fatigue, easy bruising, or more obvious bleeding, dyspnea, dizziness, bone or joint pain, anorexia, or abdominal pain. Fever, night sweats, or weight loss can occur (12,13,14,15). The total leukocyte count can be very high. Evidence of bone marrow failure is common, manifested by anemia, granulocytopenia, or thrombocytopenia. Lymphadenopathy is common; approximately half of patients have a mediastinal mass. Hepatosplenomegaly is more common in pre-T ALL patients compared with pre-T LBL patients.
The clinical course of patients with pre-T ALL/LBL is very aggressive (12,16,17). Therapy for these patients is complex, involving intensive multiagent chemotherapy regimens and prophylaxis of sanctuary sites. Usually, therapy is administered in phases: induction, consolidation, and maintenance—the latter for up to 3 years to prevent relapse. With these regimens, up to 90% event-free survival has been reported in children with pre-T ALL/LBL (12,16,17). Prognosis is relatively worse in adults.
Histologic Findings
Lymph Node
Pre-T LBL replaces lymph nodes in a diffuse pattern (1,18,19) (Fig. 59.2). In most cases, the lymph node architecture is completely replaced. In approximately one-third of cases, residual lymphoid follicles, “naked” germinal centers, small islands of residual paracortical tissue, or patent sinuses can be present. The neoplasm commonly infiltrates the lymph node capsule and extends into perinodal adipose tissue (Fig. 59.3). Within the capsule itself, and in fibrocollagenous areas within the lymph node such as the hilum, the neoplasm can show a single-cell or linear pattern of infiltration (18). A starry-sky pattern can be present and most often is a focal finding. However, in approximately 10% of cases, a starry-sky pattern is more prominent (Fig. 59.2A) (18). In some biopsy specimens, the neoplasm can be subdivided into nodules by strands of collagen
(20) (Fig. 59.4). This should not be misinterpreted as follicle formation.
(20) (Fig. 59.4). This should not be misinterpreted as follicle formation.
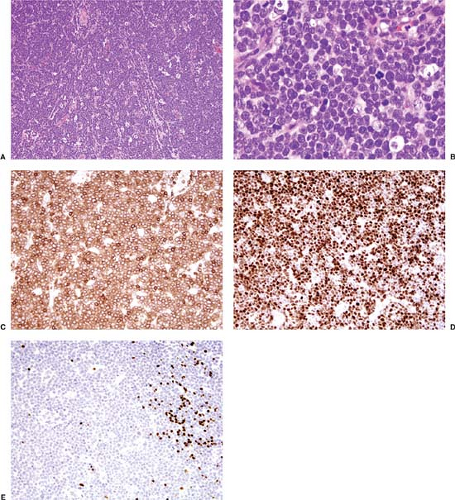 Figure 59.2. Precursor T-cell lymphoblastic leukemia/lymphoma involving lymph node. A: The neoplasm diffusely replaces the lymph node and has a starry-sky pattern. B: The neoplastic cells are small to intermediate in size with immature chromatin typical of lymphoblasts and are predominantly nonconvoluted. C–E: Immunohistochemical stains for CD3 (C), TdT (D), and PAX5/BSAP (E) supporting an immature T-cell immunophenotype. A,B, hematoxylin-eosin stain; C–E, immunohistochemistry with hematoxylin counterstain. |
Cytologically, in routinely stained tissue sections, lymphoblasts are usually smaller than the nuclei of benign histiocytes and have a high nucleus-to-cytoplasm ratio with minimal cytoplasm (1,18,19). Mitotic figures are often numerous. The chromatin of the neoplastic cells is fine and has been referred to as “dusty” or “salt and pepper.” Nucleoli are either absent or inconspicuous. The nuclear contours can be either highly irregular or round, referred to as convoluted (Fig. 59.5) or nonconvoluted (Fig. 59.2B), respectively. In most cases of pre-T LBL, cells with convoluted nuclei predominate, but usually a smaller number of nonconvoluted cells also can be identified and, in many cases, nonconvoluted cells predominate. The convoluted versus nonconvoluted nature of the lymphoblasts has no clinical significance (1).
In a subset of pre-T LBL cases, the neoplastic cells do not have the typical cytologic features just described. In some neoplasms, the cells are larger and have distinctly visible nucleoli. These neoplasms correspond to the FAB L2 category as defined in peripheral blood and bone marrow aspirate smears. Nathwani and colleagues (18) described cells that are slightly larger and more vesicular; they referred to these as prolymphocytes. These cells may overlap with L2 cytologic features. In another
subset of cases, the neoplastic cells can have coarse chromatin and resemble, in part, small cleaved cells (21).
subset of cases, the neoplastic cells can have coarse chromatin and resemble, in part, small cleaved cells (21).
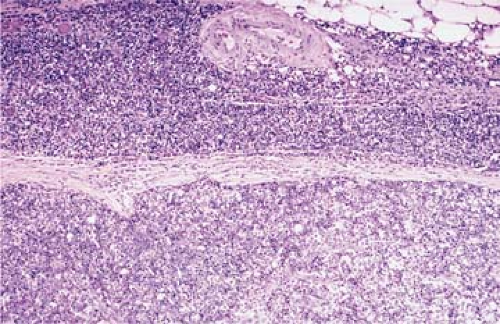 Figure 59.3. Precursor T-cell lymphoblastic leukemia/lymphoma involving lymph node. Lymphoblasts infiltrate through lymph node capsule in a single-file pattern and into perinodal adipose tissue. Hematoxylin-eosin stain. |
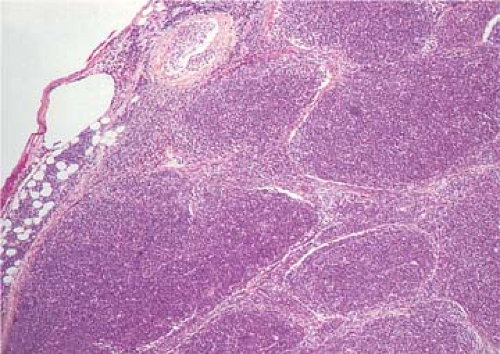 Figure 59.4. Precursor T-cell lymphoblastic leukemia/lymphoma involving lymph node. Compartmentalization of lymphoblasts by thin strands of collagen results in a pseudonodular pattern. Hematoxylin, phloxine, and saffron stain. |
 Figure 59.5. Precursor T-cell lymphoblastic leukemia/lymphoma involving lymph node. The lymphoblasts in this case are markedly convoluted. Hematoxylin-eosin stain. |
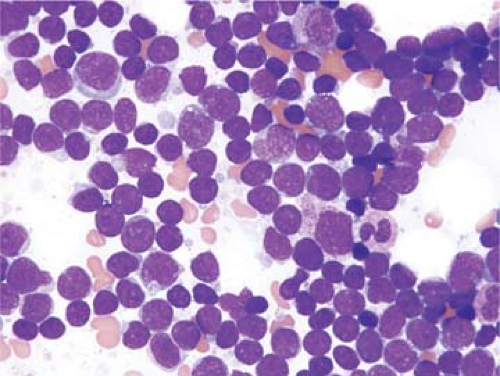 Figure 59.6. Precursor T-cell lymphoblastic leukemia/lymphoma involving bone marrow aspirate smear. In this field, the lymphoblasts have L1 features as described in the FAB classification. Wright-Giemsa stain. |
Rare cases of pre-T LBL have been reported in which the neoplastic cells are associated with either peripheral blood or tissue eosinophilia. In many of these cases, the pre-T LBL is associated with either a myeloproliferative disorder or acute myeloid leukemia, suggesting that both neoplasms arise from a common progenitor cell. One group of these patients has abnormalities of chromosome 8p11, for which the term 8p11 myeloproliferative syndrome has been coined. This syndrome is described in more detail later in this chapter. In another study, two patients who had pre-T LBL and eosinophilia were shown to carry the FIP1L1–PDGFRα fusion gene (22).
Blood and Bone Marrow
The cytologic features of pre-T ALL/LBL in blood and bone marrow aspirate smears have been well described (1,3,23). In most cases, corresponding to so-called L1 features, the lymphoblasts are small with minimal cytoplasm (Fig. 59.6). The chromatin is fine, and usually one, nonprominent nucleolus is present. In so-called L2 cases, lymphoblasts are larger, with more prominent cytoplasm and a more prominent nucleolus. Cytoplasmic vacuoles can be present, although they are usually not numerous. Lymphoblasts with cytoplasmic pseudopods, some of which are prominent and can resemble a hand mirror (so-called “hand-mirror cells”), have been described in a small subset of pre-T ALL cases; hand-mirror cells tend to be more common in pre-B ALL (24).
Electron Microscopy
Lymphoblasts are of medium size, with smooth surfaces and no cytoplasmic processes (Fig. 59.7) (25,26). The scarce cytoplasm includes few mitochondria, strands of endoplasmic reticulum, free ribosomes, and perinuclear filaments and microtubules. Cells with filament bundles are common in blood but rare in lymph nodes. The nuclei are irregularly shaped, with frequent fissures and convolutions, but usually without the serpentine or cerebriform configurations that characterize Sézary cells (25). The chromatin is evenly dispersed, with peripheral condensation. The nucleoli are dense and adjacent to the nuclear membrane.
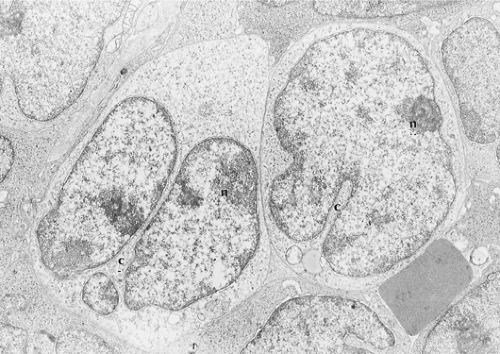 Figure 59.7. Electron micrograph of precursor T-cell lymphoblastic leukemia/lymphoma involving lymph node. Lymphoblasts have a high nucleus-to-cytoplasm ratio, a small amount of cytoplasm (c) with few organelles, and convoluted nuclei with deep indentations that sometimes create the appearance of multinucleated cells (left); the chromatin is fine with peripheral nucleoli (n). × 16,000. |
Cytochemistry
In approximately 75% of cases of pre-T ALL/LBL staining with periodic acid–Schiff (PAS) shows a discrete, block-like pattern (27). T-cell lymphoblasts also exhibit a positive dot-like acid phosphatase reaction, or a fluoride-resistant nonspecific esterase reaction in the paranuclear Golgi zone (28). Neither of these tests is specific or adequately sensitive for routine diagnosis, and these tests are infrequently used in current practice. T-cell lymphoblasts are negative for myeloperoxidase and Sudan black B (27).
Immunophenotypic Findings
During normal T-cell development, lymphoid precursors migrate from the bone marrow to the thymus (29). Within the thymus, T cells undergo T-cell receptor (TCR) gene rearrangement and selection, during which self-reactive T cells are eliminated. These processes are accompanied by the acquisition of cell surface antigens in a sequential manner. Once mature, T cells migrate from the thymus to peripheral lymphoid organs where they encounter antigen and participate in cell-mediated immunity.
As normal T cells develop, antigens are initially expressed in the cell cytoplasm and subsequently on the cell surface. CD7 is the first T-cell–associated antigen expressed on the cell surface of these neoplasms, followed by CD2 and CD5. Although cytoplasmic CD3 can be detected early in maturation, expression of this antigen on the cell surface occurs much later in maturation; this is also true for surface expression of TCR α/β or TCR γ/δ. The intensity of surface CD3 and TCRs also correlates with maturation, being dim in more immature cases. Earliest T-cell precursors are negative for both CD4 and CD8; subsequently, both antigens may be coexpressed before T-cells become either CD4+/CD8– or CD4–/CD8+. CD1a is expressed by immature T cells, but is usually not expressed by T cells at the early or late stages of maturation.
The immunophenotypes of pre-T lymphoblastic neoplasms closely reflect stages of normal T-cell maturation and appear to be “frozen” at a given stage (1,8,30). Immunophenotypic stages of normal T-cell development have been grouped in slightly different ways by various investigators. The system of the European Group for the Immunological Characterization of Leukemias (EGIL) is one system commonly used (30). The EGIL has divided the immunophenotypes of pre-T ALL/LBL into four stages corresponding to normal T-cell maturation: pro-T, pre-T, cortical (CD1a+), and mature T (30). CD7 is the earliest T-cell antigen expressed, and its presence without other T-cell antigens corresponds to the pro-T stage. In the pre-T stage, CD2 or CD5 is present as well as CD7. The acquisition of CD1a marks transition to the cortical stage, and mature T cells express multiple T-cell antigens including surface CD3 and are CD1a–. The immunophenotypes of pre-T lymphoblastic neoplasms also correlate, in part, with clinical presentation of disease. In general, neoplasms presenting as ALL tend to
have an earlier (more immature) T-cell immunophenotype than neoplasms presenting as LBL (8,13,30). This difference has not been correlated with distinct genetic abnormalities and, in most studies, has not had an impact on prognosis.
have an earlier (more immature) T-cell immunophenotype than neoplasms presenting as LBL (8,13,30). This difference has not been correlated with distinct genetic abnormalities and, in most studies, has not had an impact on prognosis.
Lymphoblasts express the enzyme terminal deoxynucleotidyl transferase (TdT). This enzyme functions physiologically by adding extra nucleotides to the joining regions between antigen receptor gene segments during the gene rearrangement process, thereby increasing genetic diversity (see Chapter 8). Terminal deoxynucleotidyl transferase is an extremely useful marker of lymphoblastic neoplasms, being positive in over 90% of all pre-T ALL/LBL (31,32). However, TdT expression is not lineage specific, as it is also expressed by over 95% of pre-B ALL/LBL, a subset of acute myeloid leukemia (mostly undifferentiated or poorly differentiated neoplasms), blastic NK cell neoplasms, and CD4+/CD56+ blastic hematodermic neoplasms. As TdT is not a cell surface antigen, if flow cytometry is used for its assessment, cell membranes must first be permeabilized before staining can be performed.
If fresh tissue is available, flow cytometry is the best approach for immunophenotyping pre-T lymphoblastic neoplasms. The ability to assess multiple antigens simultaneously and the quantitative nature of the results provide the best opportunity to exclude the possibility of a biphenotypic neoplasm. The high sensitivity of this technique also allows detection of dim antigen expression that is often not detected if one is using immunohistochemistry. Furthermore, some antigens of interest are typically not assessed in routinely processed tissue sections, most often because the antibodies are not reliably reactive.
Many cases of pre-T ALL/LBL express CD34, but this antigen may be expressed dimly and therefore is best assessed by flow cytometry (33). Many cases of pre-T ALL/LBL at an early stage of maturation are CD34+. Although most cases are CD45+, this marker is expressed relatively late in maturation and therefore pre-T lymphoblastic neoplasms corresponding to the earliest stages of maturation can be negative. This is important to remember because one of the commonly used gating strategies uses CD45 expression and side scatter, and this is a potential pitfall in analysis. Most cases of pre-T ALL/LBL are positive for HLA-DR. CD10 is expressed in a subset of cases (up to 50% to 60% in some studies), but CD10 is not expressed at the earliest or latest stages of maturation (34,35). CD4 and CD8 are coexpressed in approximately 25% of cases (36).
CD52 is expressed by virtually all pre-T ALL/LBL cases and CD38 and CD71 (transferrin receptor), are positive in a substantial subset of cases (32,37). Pre-T ALL/LBLs, particularly those arising in older adults, also can express the myeloid-associated antigens CD13 and CD33; this occurrence does not impact on presenting features or prognosis (1,7,38). C-kit (CD117) can be rarely positive in pre-T lymphoblastic neoplasms (33). Pre-T ALL/LBLs are negative for surface immunoglobulin (Ig); the pan–B-cell antigens CD19, CD20, and CD22; CD25 (<10% can be positive), CD74, CD80, and the NK-associated antigens CD16, CD57, CD94; and CD158 (killer Ig-like receptors; KIRs) (37).
In some cases, fresh tissue or bone marrow aspirate material is not available, and flow cytometry cannot be performed. In this circumstance, immunohistochemical staining of routinely processed tissue sections is also helpful in the evaluation of pre-T lymphoblastic leukemia/lymphomas. A potential advantage of this approach is that one can observe both cytoplasmic and surface expression. If one compares immunohistochemical data with the results of routine surface immunophenotyping by flow cytometry, the potential for disagreement arises. In particular, cases of pre-T ALL/LBL negative for surface CD3 shown by flow cytometry are commonly positive for cytoplasmic CD3 shown by immunohistochemistry (35). This is expected based on the normal maturation sequence, with CD3 being present in the cytoplasm prior to surface expression.
Based on their immunoreactivity in routinely processed tissue sections, certain antibodies that are infrequently used in flow cytometry panels are used in immunohistochemistry panels. CD99, best known for its expression in Ewing sarcoma/peripheral neuroectodermal tumor, is positive in approximately 80% of pre-T lymphoblastic neoplasms (34). CD43 is positive in most cases, and CD45RO in approximately half of pre-T ALL/LBL cases (35,39). The βF-1 antibody, reactive with an epitope of the TCR α/β, was positive in 36% of neoplasms in one study (35). Antiapoptotic proteins, such as BCL-2, are positive in most cases of pre-T ALL/LBL (40). MIB-1 (Ki-67) shows a variably high proliferation rate, in the range of 50% to 90% (40). Approximately 50% to 60% of cases can express the B-cell marker CD79a (33,41).
Cytogenetics Findings
Most cases analyzed by conventional cytogenetics or molecular methods have been pre-T ALL. Because pre-T LBL is under-represented, and it seems reasonable to believe that some differences may exist between cases that present as leukemia or lymphoma, it must be recognized that some bias may exist in the cytogenetic and molecular literature on pre-T lymphoblastic neoplasms.
Using conventional cytogenetics methods, approximately 25% to 50% of cases of pre-T ALL/LBL have chromosomal aberrations (Table 59.1) (29,42). Structural changes, particularly translocations, are far more common than numerical abnormalities. The frequency of abnormalities is likely to be higher using fluorescence in situ hybridization (FISH) analysis. For example, Cauwelier and colleagues (43) showed breakpoints involving the TCR β or TCR α/δ gene loci alone in approximately 40% of pre-T ALL, some of which were not detectable by conventional cytogenetic analysis.
Two types of translocations have been detected in cases of pre-T ALL/LBL (29,44). In the first type, intact genes are juxtaposed with the TCR antigen receptor gene loci, resulting in gene overexpression. In the second type, translocations or cryptic deletions result in the creation of novel fusion genes. The most common translocations of the first type involve the hox family
(44). The t(7;10)(q34;q24) and t(10;14)(q24;q11) juxtapose hox11 (also known as tlx1) with either the TCR β or TCR α/δ. The t(5;14)(q35;q32) juxtaposes hox11l2 (also known as tlx3) with bcl11b. The inv(7)(p15q34) juxtaposes the hoxa cluster, resulting in overexpression of HOXA10 and HOXA11, with TCR β. The LIM-domain only (LMO) genes are also involved in translocations of the first type. In the t(11;14)(p15;q11) and t(11;14)(p13;q11), lmo1 and lmo2, respectively are juxtaposed with the TCR α/β locus.
(44). The t(7;10)(q34;q24) and t(10;14)(q24;q11) juxtapose hox11 (also known as tlx1) with either the TCR β or TCR α/δ. The t(5;14)(q35;q32) juxtaposes hox11l2 (also known as tlx3) with bcl11b. The inv(7)(p15q34) juxtaposes the hoxa cluster, resulting in overexpression of HOXA10 and HOXA11, with TCR β. The LIM-domain only (LMO) genes are also involved in translocations of the first type. In the t(11;14)(p15;q11) and t(11;14)(p13;q11), lmo1 and lmo2, respectively are juxtaposed with the TCR α/β locus.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


