Follicular hyperplasia
Follicular lymphoma
Younger patients
Normal follicle density
Older patients
Increased follicle density (back to back)
Follicles vary in size and shape
Well-defined mantle zone
Cellular polarization present
Heterogeneous follicle cells
Tingible body macrophages present
Mitoses common
Follicles confined to node
Follicles homogeneous
Thin or absent mantle zone
Cellular polarization absent
Homogeneous follicle cells
Tingible body macrophages absent (usually)
Mitoses uncommon
Follicles may be seen in perinodal tissue
No atypical cells between follicles
Interfollicular regions may contain neoplastic cells
Follicle center cells (FCC) bcl-2 negative
Follicle center cells bcl-2 positive (usually)
FCC not light chain restricted
BCL2 translocation absent
FCC light chain restricted
BCL2 translocation present (usually)
Toxoplasmosis
Clinical
♦
Mostly young females (if pregnant, may result in birth defects in developing fetus)
♦
Transmitted by exposure to oocysts in cat feces or by ingestion of poorly cooked meat
♦
Symptoms: flu-like or asymptomatic
♦
Site of involvement: posterior cervical nodes most common
Microscopic (Fig. 15.1)
♦
Toxo triad
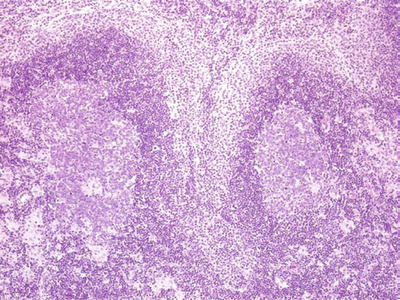
Fig. 15.1.
Toxoplasmosis lymphadenitis.
Florid follicular hyperplasia
Monocytoid B-cell hyperplasia expanding and surrounding sinuses
Paracortical epithelioid histiocyte clusters that encroach on germinal centers
•
No necrosis
♦
Confirm diagnosis with serologic studies
Cytomegalovirus
Microscopic
♦
Florid follicular hyperplasia
♦
Monocytoid B-cell hyperplasia expanding sinuses (CMV inclusions may sometimes be identified here)
♦
+/− immunoblastic proliferation (may be atypical)
♦
Confirm diagnosis serologically, immunohistochemically, or by in situ hybridization techniques
Human Immunodeficiency Virus
Microscopic (Fig. 15.2)
♦
Early stage
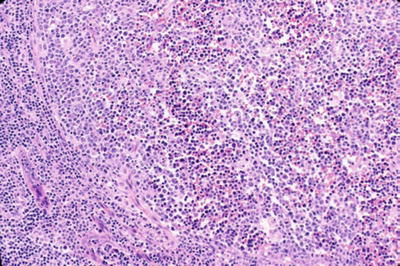
Fig. 15.2.
Human immunodeficiency virus lymphadenitis.
Florid reactive lymphoid hyperplasia with absent mantle zones and follicle lysis of germinal centers (germinal centers disrupted by hemorrhage, disrupted FDC [follicular dendritic cell] meshwork, and increased T cells)
Monocytoid B-cell hyperplasia expanding sinuses
Epithelioid histiocyte clusters
Increased plasma cells and polykaryocytes (large multinucleated giant cells) in interfollicular zones
♦
Late stage
Regressively transformed germinal centers
Depletion of lymphocytes from paracortex
Rheumatoid Arthritis
Microscopic
♦
Florid follicular hyperplasia
♦
Marked interfollicular plasmacytosis (plasma cells also present within follicles)
♦
Clusters of neutrophils in sinuses
Differential Diagnosis
♦
Other inflammatory disorders (e.g., Sjögren syndrome, Felty syndrome, Still disease)
♦
HIV infection
♦
Syphilis
♦
Castleman disease, plasma cell type
Syphilis (Luetic lymphadenitis)
Microscopic
♦
Florid follicular hyperplasia
♦
Interfollicular plasmacytosis
♦
Epithelioid granulomas
♦
Thick, fibrotic capsule – perivascular plasma cells
♦
+/− arteritis/phlebitis
♦
Confirm diagnosis serologically and/or immunohistochemically
Progressive Transformation of Germinal Centers
♦
Rarely precedes, accompanies, or follows nodular lymphocyte predominant Hodgkin lymphoma (NLPHL)
Microscopic
♦
Associated with reactive follicular hyperplasia in the same node
♦
Large follicles with indistinct germinal center/mantle zone borders due to infiltration of germinal centers by mantle zone lymphocytes
♦
Residual centroblasts may mimic LP cells of NLPHL
Differential Diagnosis
♦
NLPHL
Neoplastic Hodgkin cells have nuclear lobulation, while centroblasts usually do not
♦
Follicular lymphoma, floral variant
Neoplastic follicle center cells are usually bcl-2+ and exhibit kappa or lambda light chain restriction
♦
Mantle cell lymphoma
Neoplastic cells surround atrophic follicles
Monoclonal B cells: CD5+, cyclinD1+
Castleman Disease: Hyaline Vascular Type
Clinical
♦
Usually solitary mediastinal mass, may involve other sites
Microscopic (Fig. 15.3)
♦
Atrophic germinal centers (regressively transformed) with expanded mantle zones composed of concentric layers of lymphocytes
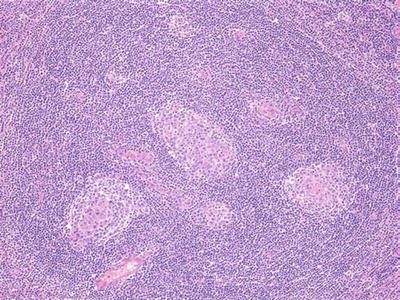
Fig. 15.3.
Castleman disease, hyaline vascular type.
♦
Multiple regressively transformed germinal centers in one “cloud” of mantle zone lymphocytes
♦
Hyalinized blood vessels penetrate into follicles; interfollicular vascularity increased
♦
Few interfollicular plasma cells or transformed lymphocytes
Differential Diagnosis
♦
HIV infection
Absent mantle zones
I ncreased interfollicular plasma cells and polykaryocytes
Castleman Disease: Plasma Cell Variant
Clinical
♦
Patients may have POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin lesions) or a variety of other signs and symptoms
♦
Sites of involvement: abdominal cavity and mediastinum, peripheral lymph nodes, extranodal sites
♦
Clinicopathologic diagnosis
Microscopic
♦
Florid follicular hyperplasia with regressive transformation of germinal centers
♦
Marked interfollicular plasmacytosis that extends to the lymph node capsule
♦
When multicentric, may be HHV8-positive
Differential Diagnosis
♦
Rheumatoid arthritis
♦
HIV infection
♦
Syphilis
Sinus Hyperplasias
♦
Differential diagnosis of all sinus hyperplasias
Metastatic carcinoma
Metastatic malignant melanoma
Anaplastic large-cell lymphoma (ALCL)
Whipple Disease
Clinical
♦
Age: middle -aged adults
♦
Sex: M > F
♦
Etiology: infectious disease of small bowel (etiologic agent: Tropheryma whipplei, formerly called Tropheryma whippelii )
♦
Site of involvement: small bowel, abdominal lymph nodes, +/− peripheral lymph nodes
Microscopic
♦
Sinuses distended by large lipid vacuoles surrounded by vacuolated histiocytes
♦
PAS+ bacilli present within histiocyte cytoplasm in sinuses and in germinal centers
♦
Confirm diagnosis by PCR or electron microscopy
Differential Diagnosis
♦
Mycobacterium avium-intracellulare (MAI): organisms are acid-fast + as well as PAS+
♦
Lymphangiogram effect: histiocytes are PAS– and acid-fast –
♦
Lipogranuloma: diagnosis of exclusion
♦
Silicon
Hemophagocytic Syndrome
Clinical
♦
Immunocompromised host
♦
Usually due to viral infection, may result from bacterial, fungal, or parasitic infection
♦
May complicate certain lymphomas
Subcutaneous panniculitis-like T-cell lymphoma
ALCL
Extranodal NK-/T-cell lymphoma, nasal type
♦
Sites of involvement: lymph nodes, spleen, bone marrow
Microscopic
♦
Sinuses distended by benign-appearing histiocytes containing phagocytized erythrocytes or other hematopoietic elements
Vascular Transformation of Lymph Node Sinuses
Clinical
♦
Lymph node enlargement
♦
Sometimes associated with deep venous thrombosis in adjacent vein
Microscopic
♦
Sinuses distended by proliferating anastamosing vascular channels, often with fibrosis
♦
Capsule spared
Differential Diagnosis
♦
Kaposi sarcoma
Capsular/subcapsular involvement
Spindle cell proliferation without distinct vascular channels
PAS + hyaline globules
Paracortical Hyperplasias
♦
Differential diagnosis of all paracortical hyperplasias
Peripheral T-cell lymphoma
Interfollicular classical Hodgkin lymphoma
Infectious Mononucleosis
Microscopic (Fig. 15.4)
♦
Paracortical expansion (EBV+ B cells located in the paracortex)
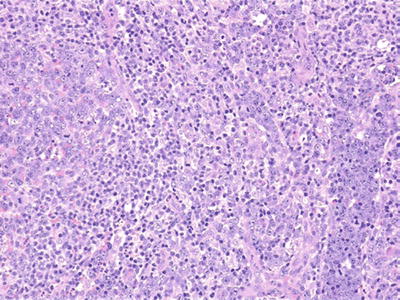
Fig. 15.4.
Infectious mononucleosis.
♦
Focal necrosis
♦
Sinuses distended by atypical lymphocytes, monocytoid B cells, and/or immunoblasts
♦
+/− follicular hyperplasia
♦
Cytology: polymorphous population of transformed lymphocytes, immunoblasts, RS-like cells, plasma cells, and histiocytes
♦
Confirm EBV+ immunoblasts by in situ hybridization (EBER) or immunohistochemistry (LMP)
♦
Confirm diagnosis by serologic studies
Differential Diagnosis
♦
Diffuse large B-cell lymphoma
♦
Classic Hodgkin lymphoma
♦
Viral infection (e.g., CMV)
♦
Drug reaction (especially hydantoin)
Atypical Immunoblastic Reaction
Microscopic
♦
Similar to infectious mononucleosis
Causes
♦
Drug reaction (especially hydantoin): eosinophils often numerous
♦
Herpes simplex: viral inclusions may be seen in and around necrotic areas
Dermatopathic Lymphadenopathy
Clinical
♦
Usually associated with skin lesions
Microscopic
♦
Subcapsular paracortical regions expanded (often focally) by small lymphocytes, some with convoluted nuclei resembling small Sézary cells, interdigitating reticulum cells, Langerhans cells, and histiocytes containing melanin, lipid, and hemosiderin
♦
Residual node displaced centrally
Differential Diagnosis
♦
Mycosis fungoides
More architectural effacement, large- and medium-sized pleomorphic lymphocytes present
Aberrant T-cell phenotype may be present
Necrotizing Granulomatous Lymphadenitis
Cat Scratch Disease
Clinical
♦
Sites of involvement: axillary and cervical lymph nodes
♦
Etiologic agent: Bartonella henselae
Microscopic
♦
Central stellate abscesses containing neutrophils, surrounded by palisaded histiocytes and fibroblasts ; sparse to no multinucleated giant histiocytes
♦
+/− follicular hyperplasia
♦
+/− monocytoid B cells distending sinuses
♦
Bacilli (found in necrotic areas) stain positively with Warthin–Starry stain
♦
Diagnosis can be confirmed by PCR
Differential Diagnosis
♦
Lymphogranuloma venereum (LGV), tularemia, Yersinia
All negative on Warthin–Starry stain
♦
Toxoplasmosis: no necrosis, Warthin–Starry negative
♦
Hodgkin lymphoma
Lymphogranuloma Venereum
Clinical
♦
Sexually transmitted disease caused by Chlamydia trachomatis
♦
Involves inguinal lymph nodes in males and pelvic lymph nodes in females
Microscopic
♦
Morphologically indistinguishable from cat scratch disease, tularemia, and Yersinia
♦
Confirm diagnosis serologically
Tularemia
♦
Often history of tick bite
♦
Involves axillary lymph nodes
♦
Morphologically indistinguishable from cat scratch disease, LGV, and Yersinia
♦
Confirm diagnosis by serology or cultures
Yersinia
♦
Clinical history of abdominal pain and diarrhea, signs suggesting appendicitis
♦
Involves mesenteric lymph nodes
♦
Morphologically indistinguishable from cat scratch disease, LGV, and tularemia
Tuberculosis
Microscopic
♦
Necrotizing granulomas with central caseous necrosis without neutrophils
♦
AFB + bacilli present in necrotic areas (may be difficult to find); PCR may help
Fungal Infection
♦
Morphologically similar to TB, but granulomas more commonly contain neutrophils and karyorrhectic nuclear debris
♦
Organisms: GMS+, PAS+
Necrotizing Nongranulomatous Lymphadenitis
Kikuchi–Fujimoto Disease
Synonym
♦
Histiocytic necrotizing lymphadenitis
Clinical
♦
Young adults females
♦
Involves cervical lymph nodes
Microscopic (Fig. 15.5)
♦
Patchy cortical and paracortical necrosis with extensive karyorrhectic debris and histiocytes centrally
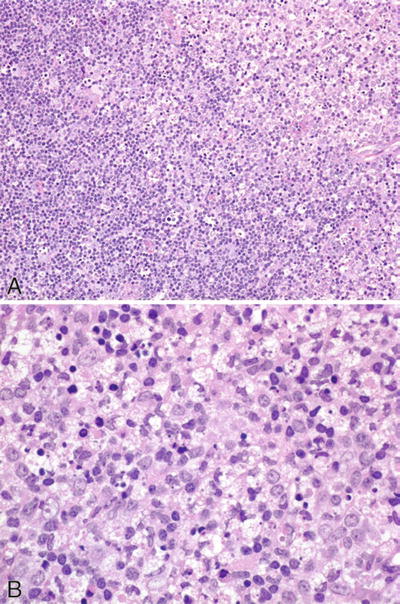
Fig. 15.5.
Kikuchi–Fujimoto disease (A, B).
♦
Immunoblasts, histiocytes, and plasmacytoid monocytes peripherally
♦
No granulocytes, few plasma cells
Systemic Lupus Erythematosus
Clinical
♦
Young adults females
♦
Cervical or generalized lymphadenopathy
Microscopic
♦
Follicular hyperplasia
♦
Interfollicular zones contain increased plasma cells and immunoblasts
♦
Areas of necrosis may contain neutrophils or plasma cells
♦
Hematoxylin bodies (composed of DNA aggregates, polysaccharides, and immunoglobulin) found within areas of necrosis as well as in walls of blood vessels
Malignant Lymphoma/Leukemia (WHO Classification)
Precursor B- and T-Cell Neoplasms
B-Lymphoblastic Leukemia/Lymphoma, Not Otherwise Specified
Clinical
♦
Age: children > adults
♦
Presentation
Acute lymphoblastic leukemia (bone marrow and peripheral blood involvement ) – more common. Pancytopenia, adenopathy, hepatosplenomegaly, bone pain
Lymphoblastic lymphoma (solid tumor [lymph node, skin, bone], +/− minimal bone marrow, or peripheral blood involvement) – less common. Mediastinum rarely involved in B-LBL
♦
Clinical course
Highly aggressive
Often curable with chemotherapy
♦
Postulated cell of origin
Precursor B lymphoblasts
Microscopic
♦
Low power (lymph node)
Architecture effaced
Invasion of perinodal fat common
Tumor involves paracortex, may spare reactive follicles
♦
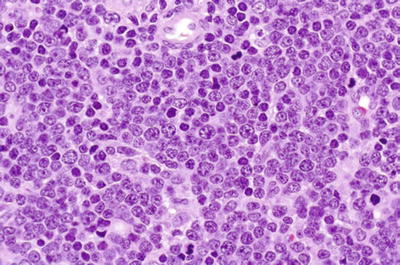
Monotonous population of lymphoblasts: medium-sized cells with round or convoluted nuclei, fine chromatin, inconspicuous nucleoli, scant cytoplasm
Frequent mitoses
+/− starry-sky pattern (tingible body macrophages)
T and B phenotypes morphologically indistinguishable
Immunophenotype
♦
CD19+, CD79a+
♦
CD43+
♦
TdT (terminal deoxynucleatidyl transferase) +
♦
CD34 usually +
♦
CD20, CD22 usually +
♦
CD10 usually +
♦
Surface immunoglobulin (sIg) –
♦
CD13 and/or CD33 may be present
Genetics
♦
Ig heavy chain genes rearranged
♦
Light chain genes may be rearranged (~50%)
♦
T-cell receptor gene may be rearranged in B-LBL
Differential Diagnosis (Table 15.2)
♦
More mature B-cell neoplasms (e.g., blastoid variant of mantle cell lymphoma)
Table 15.2.
Paraffin Section Immunophenotype of Blastic Hematolymphoid Malignancies
Disorder | TdT | CD34 | CD43 | MPO/Lys | CD3 | CD79a | CD20 | Cyclin D1 |
|---|---|---|---|---|---|---|---|---|
T-lymphoblastic leukemia/lymphoma | + | – | + | − | + | − | – | – |
B-lymphoblastic leukemia/lymphoma | + | + | + | – | – | + | +/− | − |
Myeloid sarcoma | +/− | +/− | + | + | − | − | – | – |
Burkitt lymphoma | − | − | + | − | − | + | + | − |
Blastoid mantle cell lymphoma | − | − | + | − | − | + | + | + |
TdT and CD34–
sIg+
♦
T-lymphoblastic leukemia/lymphoma
B-cell-associated antigens negative
CD3, CD7+
CD34 negative
♦
Myeloid sarcoma
CD13, CD33, myeloperoxidase (MPO), CD68, lysozyme +
CD19, CD22, CD10, CD79a negative
♦
Burkitt lymphoma
More prominent starry-sky pattern, coarser chromatin, multiple nucleoli, amphophilic, granular cytoplasm
sIg+
CD34, TdT negative
B-Lymphoblastic Leukemia/Lymphoma with Recurrent Genetic Abnormalities (common variants)
♦
B-lymphoblastic leukemia/lymphoma with t(9;22)(q34;q11.2); BCR-ABL1
More common in adults than children
Poor prognosis
♦
B-lymphoblastic leukemia/lymphoma with t(v;11q23); MLL rearranged
Usually occurs in infants
Poor prognosis
♦
B-lymphoblastic leukemia/lymphoma with t(12;21)(p13;q22); TEL-AML1 (ETV6-RUNX1)
Common in children, rare in adults
Very favorable prognosis
♦
B-lymphoblastic leukemia/lymphoma with hyperdiploidy
Common in children, rare in adults
Very favorable prognosis
♦
B-lymphoblastic leukemia/lymphoma with t(1;19)(q23;p13.3); E2A-PBX1 (TCF3-PBX1)
Relatively common in children
CD19+, CD10+, cytoplasmic μ heavy chain-positive pre-B-cell phenotype
T-Lymphoblastic Leukemia/Lymphoma
Clinical
♦
Age: adolescents and young adults
♦
Sex: M > F
♦
Presentation : rapidly enlarging mediastinal (thymic) mass, +/− lymphadenopathy, SVC syndrome, pericardial and pleural effusions
♦
Bone marrow involvement >25% = precursor T-lymphoblastic leukemia
♦
Clinical course: highly aggressive but potentially curable
Microscopic (Fig. 15.7)
♦
Monotonous population of lymphoblasts (medium-sized cells with round nuclei, fine chromatin, inconspicuous nucleoli, and scant cytoplasm)
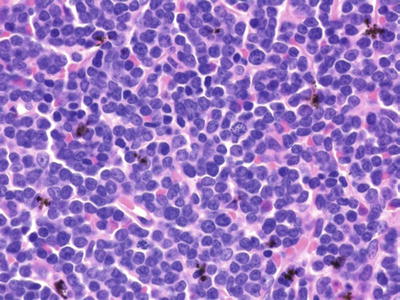
Fig. 15.7.
T-lymphoblastic leukemia/lymphoma.
♦
Numerous mitoses, tingible body macrophages (starry sky)
♦
Morphologically indistinguishable from B-LBL
Immunophenotype
♦
Mirrors stages of intrathymic T-cell ontogeny
♦
CD7+, CD3+ (cytoplasmic + even if surface –), CD2+, CD5+
♦
Subset of cases expresses CD1, CD4, and CD8
♦
TdT+, CD34+/-, CD1a+, CD99+
♦
Immunoglobulin negative
♦
B-cell-associated antigens negative, although CD79a may be +
♦
May be positive for CD13 or CD33
Genetics
♦
T-cell receptor gene rearrangement variable
♦
Immunoglobulin heavy chain gene rearrangement may be present
Differential Diagnosis
♦
B-lymphoblastic leukemia/lymphoma
B-cell-associated antigens+
T-cell-associated antigens negative
CD10+
♦
Mature B-cell lymphoma
sIg+
B-cell-associated antigens+
T-cell-associated antigens negative
TdT negative
♦
Mature T-cell lymphoma
Heterogeneous cell population that lacks blastic cytology
TdT negative
♦
Myeloid sarcoma
Eosinophilic cytoplasm
Myeloperoxidase (MPO)+, lysozyme+, CD68+
T-cell-associated antigens negative
Mature B-Cell Neoplasms
Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
Clinical
♦
Age: older adults (median age 60–65 years)
♦
Sex: M > F (slight)
♦
History of waxing and waning adenopathy common
♦
SLL and CLL morphologically indistinguishable on lymph node biopsy
♦
Clinical course
Indolent but not curable with available therapy
Median survival: 60–70 months
May transform to diffuse large B-cell lymphoma (DLBCL); poor prognosis; risk increases over time
May also develop de novo DLBCL ; better prognosis than transformation
Postulated cell of origin
•
40–50% – naïve B cell (germline variable region IG genes)
•
50–60% – postgerminal center B cell (variable region IG genes contain somatic mutations)
Poorer prognosis
•
Extensive clinical disease
•
Low ECOG performance status
•
Increased number of large cells (prolymphocytes and paraimmunoblasts)
•
17p deletion, 11q deletion
•
Germline IGHV genes
•
Elevated LDH
•
Elevated serum β2-microglobulin level
Better prognosis
•
Low stage
•
High ECOG performance status
•
Mutated IGHV genes
•
Isolated 13q deletion
•
Lack of expression of CD38/ZAP-70 (controversial)
Microscopic
♦
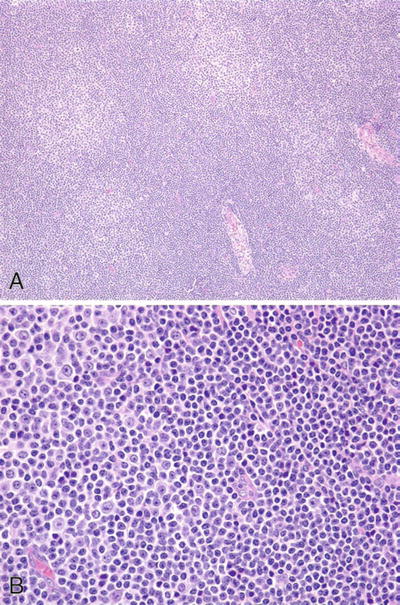
Low power
Effaced architecture although residual follicles may be present
Pale zones (proliferation centers) alternating with darker zones (Fig. 15.8A)
Fig. 15.8.
Chronic lymphocytic leukemia/small lymphocytic lymphoma. (A) Low-power and (B) high-power proliferation centers.
♦
High power
Monotonous population of small lymphocytes (rounded nuclei, clumped chromatin, scant cytoplasm, inconspicuous to small nucleoli, and infrequent mitotic figures) interspersed by scattered proliferation centers (indistinct nodules of prolymphocytes and paraimmunoblasts ) (Fig. 15.8B)
Immunophenotype
♦
CD19, CD20 (weak), CD79a+
♦
CD23+
♦
CD5+, CD43+
♦
sIg weakly + (M + / − D), immunoglobulin light chain restricted
♦
CD10–
♦
Cyclin D1–
Genetics
♦
Ig heavy and light chain genes are rearranged
♦
NOTCH1 mutation (10–15%)
♦
SF3B1 mutation (10–15%)
♦
Trisomy 12 – common – intermediate prognosis
♦
13q abnormalities (13q14) – most common – slightly better prognosis if sole abnormality
♦
11q deletion – poorer prognosis
♦
17p deletion – poorer prognosis
♦
Mantle cell lymphoma (MCL)
Table 15.3.
Morphologic Differential Diagnosis of B-Cell Lymphoproliferative Disorders in Lymph Node, Spleen, and Bone Marrow Histologic Sections
Lymph node | Spleen | Bone marrow | Cytology | |
|---|---|---|---|---|
Chronic lymphocytic leukemia/small lymphocytic lymphoma | Diffuse pattern, proliferation centers | Red pulp cords and sinusoids | Intertrabecular nodules and interstitial | Small lymphocytes, prolymphocytes, and paraimmunoblasts |
Lymphoplasmacytic lymphoma | Diffuse pattern | Red pulp cords and sinusoids, encroaches on borders of white pulp | Intertrabecular nodules and interstitial, may be paratrabecular | Small lymphocytes, plasmacytoid lymphocytes, plasma cells, Dutcher bodies |
Mantle cell lymphoma | Diffuse or nodular pattern with atrophic germinal centers | White pulp with atrophic germinal centers and obliterated marginal zones | Intertrabecular nodules and paratrabecular aggregates | Monotonous small lymphocytes with nuclear irregularity. No large cells |
Nodal marginal zone lymphoma | Perisinusal or surrounding benign germinal centers and mantle zones | White pulp with small germinal centers and residual nonneoplastic mantle cells | Intertrabecular nodules and paratrabecular aggregates | Medium-sized cells with irregular nuclei, abundant pale cytoplasm. Occasional large transformed cells |
Hairy cell leukemia | Rarely involves lymph node; hilum and perinodal soft tissue only | Commonly involved; red pulp cords and sinusoids; blood lakes | Commonly involved; interstitial; may be subtle | Small- to medium-sized lymphocytes with abundant pale cytoplasm |
Follicular lymphoma | True follicular nodularity | White pulp germinal centers expanded with benign mantle and marginal zone cells | Intertrabecular nodules and paratrabecular aggregates | Small cleaved cells and large noncleaved cells in varying proportions |
Table 15.4.
Immunophenotypic Features of Small B-Cell Lymphoproliferative Disorders
Slg | CD19 | CD20 | CD23 | CD10 | CD5 | CyclinD1 | |
|---|---|---|---|---|---|---|---|
Chronic lymphocytic leukemia/small lymphocytic lymphoma | Monoclonal (dim) | + | + (dim) | + | – | + | – |
Lymphoplasmacytic lymphoma | Monoclonal cIg in plasma cell | + | + | −/+ | – | −/+ | – |
Mantle cell lymphoma | Monoclonal (bright) | + | + (bright) | – | – | + | + |
Marginal zone lymphoma | Monoclonal (bright) | + | + (bright) | – | – | −/+ | – |
Hairy cell leukemiaa | Monoclonal (bright) | + | + | – | – | – | + (weak) |
Follicular lymphoma | Monoclonal (bright) | + | + (bright) | −/+ | + | – | – |
Slightly larger lymphocytes with cleaved nuclei; no proliferation centers
CD23 negative
Cyclin D1+
t(11;14)(q13;q32) usually present
♦
Lymphoplasmacytic lymphoma
Monoclonal plasmacytoid lymphocytes and plasma cells present
Proliferation centers usually absent
♦
Follicular lymphoma
Follicular pattern, no proliferation centers
CD5 negative, CD43 negative
CD10+, often with aberrant bcl-2 expression
Cyclin D1–
t(14;18)(q32;q21) common (although also has rarely been identified in CLL/SLL)
♦
Marginal zone lymphoma
Neoplastic lymphocytes have a monocytoid appearance, no proliferation centers
CD5 usually negative, CD23 negative, CD10 negative
♦
T-cell prolymphocytic leukemia
T-cell-associated antigens +
B-cell-associated antigens negative
♦
B-cell prolymphocytic leukemia (see below)
B-Cell Prolymphocytic Leukemia
Clinical
♦
Often present with higher WBC count and splenomegaly
Microscopic
♦
65% of blood lymphocytes are prolymphocytes (clumped chromatin with prominent single nucleolus; defined on the basis of peripheral blood involvement, not tissue sections)
Immunophenotype
♦
Strong sIg+, CD5 may be negative, CD23 usually absent
Genetics
♦
Abnormalities involving MYC (translocations and/or extra copies) common
Prognosis
♦
More aggressive clinical course
Lymphoplasmacytic Lymphoma/Waldenström Macroglobulinemia
Clinical
♦
Age: older adults (median 63 years)
♦
Sex: M > F (slight)
♦
Waldenström macroglobulinemia (WM): clinical diagnosis characterized by monoclonal IgM serum paraprotein , bone marrow-based disease, and +/– hyperviscosity signs/symptoms; pathologic diagnosis is lymphoplasmacytic lymphoma (LPL)
♦
Lymphoplasmacytic lymphoma may present with lymphadenopathy without bone marrow involvement; clinically not Waldenström macroglobulinemia; may still have monoclonal immunoglobulin serum or urine (usually IgM, occasionally IgG or IgA)
♦
Autoimmune hemolytic anemia, autoimmune thrombocytopenia, coagulation factor inhibitors, and cryoglobulinemia may be present
♦
Sites of involvement
Lymph nodes, bone marrow, spleen (usually stage III or IV at presentation)
Peripheral blood lymphocytosis may be present
♦
Clinical course
Similar to CLL/SLL
Indolent but not curable with available therapy
May transform to diffuse large B-cell lymphoma (~10%) – poor prognosis
Microscopic (Fig. 15.9)
♦
Diffuse or parafollicular proliferation of small lymphocytes, plasmacytoid lymphocytes (lymphocytic-like nuclei, plasma cell-like cytoplasm), and plasma cells
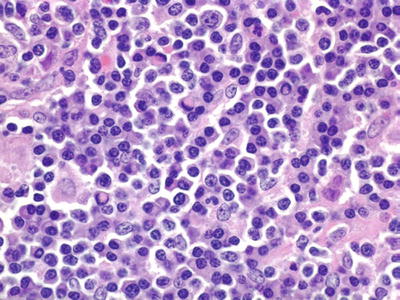
Fig. 15.9.
Lymphoplasmacytic lymphoma.
♦
Abnormal immunoglobulin production manifested by:
Dutcher bodies (intranuclear immunoglobulin inclusions)
Russell bodies (extracellular hyaline immunoglobulin bodies)
Crystalline immunoglobulin deposits (intracytoplasmic or extracellular)
Amyloid deposits
Intercellular light chain deposits, mimicking amyloid
♦
Sinuses spared (may contain immunoglobulin)
♦
Mast cells frequently seen
♦
Iron-containing epithelioid histiocytes may be present
Immunophenotype
♦
Small lymphocytes, plasmacytoid lymphocytes, and plasma cells are all light chain restricted
♦
sIg present (usually IgM; IgD absent), monoclonal cIg in plasma cells
♦
By flow cytometry, the monoclonal CD38+/CD138+ plasma cells typically coexpress CD19 and CD45, while the monoclonal plasma cells of multiple myelomas are usually negative for CD19 and CD45
♦
CD19, CD20, CD22, CD79a+
♦
CD23 usually –
♦
CD43− or +
♦
CD5 usually – and occasionally +
♦
CD10 usually –
Genetics
♦
Ig heavy and light chain genes are rearranged
♦
MYD88 L265P somatic mutation : present in >90% of LPL; is also occasionally present in other small B-cell lymphomas (e.g., splenic MZL, CLL/SLL)
Differential Diagnosis
♦
CLL/SLL
Proliferation centers present
Monoclonal plasmacytoid lymphocytes and plasma cells rare/absent
CD5+, CD23+, sIg weakly +
♦
Other small B-cell lymphomas
May be difficult to distinguish LPL from marginal zone lymphoma
MYD88 mutations far more common in WM/LPL than in MZL
Absence of monoclonal plasma cells favors MZL and essentially excludes LPL
Splenic Marginal Zone Lymphoma
Clinical
♦
Age: older adults (median seventh decade)
♦
Sex: M > F
♦
Marked splenomegaly, often without adenopathy
♦
Bone marrow and peripheral blood usually involved
♦
May present as leukemia (+/− villous lymphocytes: lymphoid cells with abundant cytoplasm and small thin villi, often concentrated at one pole, as seen on peripheral blood smear)
♦
Clinical course: indolent
♦
Splenectomy may be treatment of choice (+/− chemotherapy)
♦
May transform to diffuse large B-cell lymphoma
Microscopic (Spleen)
♦
Low power
Both mantle and marginal zones involved
Residual germinal centers atrophic or hyperplastic
Red pulp may be involved – sinusoidal involvement correlates with leukemic phase
♦
High power
Biphasic pattern involving the mantle zone and marginal zone
Mantle zone: small neoplastic lymphoid cells with little cytoplasm
Marginal zone: medium-sized neoplastic cells with moderate–abundant pale cytoplasm and scant large transformed lymphocytes with round nuclei, prominent nucleoli, dispersed chromatin, and abundant cytoplasm
Plasmacytic differentiation may be present
Immunophenotype
♦
sIg+ (M > G or A), cIg may be +; light chain restricted
♦
CD19, CD20, CD22, CD79a+
♦
CD5 and CD43 usually –, CD10– (may be + on flow cytometry)
♦
May be weakly TRAP +
♦
CD23–, cyclin D1–
♦
CD11c usually +
♦
CD103–
♦
Annexin negative, BRAF negative
Genetic Features
♦
Loss of 7q31-32 common (40–50%); rare in other small B-cell lymphomas
♦
MYD88 L265P somatic mutation : present in ~10%
Differential Diagnosis
♦
Splenic marginal zone hyperplasia
Polyclonal B cells and plasma cells
♦
Hairy cell leukemia (HCL)
Red pulp process, blood lakes common, monotonous tumor cells
Immunophenotyping: CD103+, TRAP+, DBA.44+, annexin-1+, TBET+, BRAF+
Molecular genetics: BRAF mutation present
♦
CLL/SLL
Red pulp process, architecture effaced, proliferation centers present, and cells have scant cytoplasm
CD5+, CD43+, CD23+
♦
MCL
Architecture effaced, monotonous cells with scant cytoplasm
CD5+, CD43+, cyclin D1+
♦
FL
Follicular pattern, at least partially; germinal centers expanded
CD10 and bcl-6 usually +, bcl-2 usually aberrantly coexpressed
Hairy Cell Leukemia
Clinical
♦
Age: middle-aged to older adults
♦
Sex: M > F
♦
Presentation
Splenomegaly, pancytopenia, monocytopenia
Hepatomegaly variable, although the liver is usually involved
Lymphadenopathy is uncommon
♦
Peripheral blood: always involved, but characteristic hairy cells (lymphoid cells with abundant pale cytoplasm with circumferential hairy projections) may be hard to find
♦
Bone marrow: always involved, may be inaspirable due to increased reticulin fibrosis
♦
Clinical course
Complete and durable remissions common with purine analog therapy
Microscopic
♦
Spleen
Low power
•
Involves red pulp cords and sinusoids, white pulp atrophic
•
Blood lakes (variable size) lined by neoplastic cells common
High power
•
Monotonous population of small–medium lymphoid cells with oval–reniform nuclei and abundant pale cytoplasm
•
Cells appear widely spaced
♦
Lymph node (uncommonly involved)
Prominent hilar and perinodal fat involvement
Immunophenotype
♦
sIg+, light chain restricted
♦
CD19, CD20, CD22, CD79a+
♦
CD11c+ (strong), CD25+ (strong)
♦
CD103+ (most specific marker)
♦
DBA.44+, cyclin D1+ (weak) – tissue sections
♦
TRAP+
♦
Annexin-1+, TBET+, BRAF+
♦
VE1 (V600E mutation-specific antibody) +
♦
CD5–, CD10–, CD23–, cyclin D1–
Genetics
♦
Ig heavy and light chains rearranged (proves B-cell lineage)
♦
BRAF V600E mutation present in ~100%
Electron Microscopy
♦
Long microvilli with broad base and ribosome–lamella complexes
♦
Characteristic but not pathognomonic
Differential Diagnosis
♦
Splenic marginal zone lymphoma
Involves white pulp mantle and marginal zones, blood lakes absent
CD103–, TRAP weakly + in cell subset
♦
Mastocytosis
Tryptase+, CD103–, TRAP–
♦
CLL/SLL
Architecture effaced, proliferation centers present, cells have scant cytoplasm
CD5+, CD43+, CD23+
CD103–, TRAP–
♦
MCL
White pulp process, architecture effaced, monotonous cells with scant cytoplasm
CD5+, CD43+, cyclin D1+ (strong)
♦
FL
Follicular pattern, at least partially; germinal centers expanded
Involves splenic white pulp
CD10, bcl-6 usually +
CD103–, TRAP–
Extranodal Marginal Zone Lymphoma of Mucosa-Associated Lymphoid Tissue (MALT Lymphoma)
Clinical
♦
Age: adults (median age approximately 60 years; wide age range)
♦
Sex: F > M (slight)
♦
More common in patients with autoimmune disorder (e.g., Sjögren syndrome) or chronic antigenic stimulation (e.g., Helicobacter gastritis)
♦
Sites of involvement
Extranodal by definition: stomach, salivary gland, lung, thyroid, skin, etc
May secondarily involve regional lymph nodes
Bone marrow involvement uncommon (15–20%); peripheral blood involvement rare
♦
Clinical course
Indolent; when disseminated, usually incurable with available therapy
5 year survival 75–80%
May recur in other extranodal sites
May transform to large B-cell lymphoma
Microscopic (Fig. 15.10)
♦
Low power: perisinusoidal, parafollicular, or marginal zone distribution; mantle zone preserved. Tumor gradually effaces architecture, circumscribing and infiltrating germinal centers (follicular colonization)
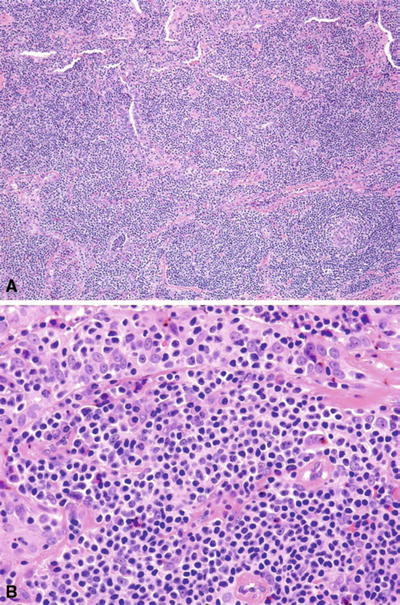
Fig. 15.10.
Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma) (A, B).
♦
High power: heterogeneous population of marginal zone cells (small–medium lymphoid cells with round slightly indented nuclei, clumped chromatin, abundant pale cytoplasm, and well-defined cytoplasmic membranes), monocytoid B cells, small lymphocytes, and plasma cells (plasma cells usually located in interfollicular zones). Occasional large transformed cells may be seen
♦
Lymphoepithelial lesions (marginal zone cells infiltrating epithelium) typically seen
♦
Plasma cells often located in subepithelium
Immunophenotype
♦
sIg+ (M>G or A)
♦
B cells light chain restricted
♦
Plasma cells may also be light chain restricted (40%)
♦
CD19, CD20, CD22, CD79a+
♦
CD5, CD43 usually negative (positive in minority of cases)
♦
CD10, CD23, cyclin D1–
♦
CD11c may be weakly +, CD25–
♦
Usually IgD–
Genetics
♦
Multiple mutually exclusive chromosomal translocations have been identified, (t(11;18)(q21;q21)/BIRC3/MALT1, t(14;18)(q32;q21)/IGH-MALT1, t(1;14)(p22;q32)/IGH-BCL10, and t(3;14)(p14.1;q32)/IGH-FOXP3); frequencies vary with site of primary disease
♦
Trisomy 3 and trisomy 18 common
Differential Diagnosis
♦
Nodal/splenic marginal zone lymphoma
Morphology and immunophenotype very similar to MALT lymphoma
MALT lymphoma’s characteristic chromosomal translocations are absent in nodal and splenic marginal zone lymphoma
♦
CLL/SLL
Architecture effaced, proliferation centers present, cells have scant cytoplasm
CD5+, CD43+, CD23+
♦
MCL
Architecture effaced; monotonous cells with scant cytoplasm
CD5+, cyclin D1+
t(11;14)(q13;q32) present
MALT lymphoma’s characteristic chromosomal translocations are absent
♦
FL
Follicular pattern, at least partially; centrocytes and centroblasts
CD10 and bcl-6 usually +, bcl-2 usually aberrantly coexpressed
♦
Monocytoid B-cell hyperplasia
Germinal centers intact – not invaded by neoplastic monocytoid cells
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


