Liver Disorders and Gallstones
This chapter looks at the chemical pathology of liver and gall bladder disorders. These are common in clinical practice, and liver function tests constitute one of the most frequently requested clinical biochemistry laboratory profiles.
FUNCTIONS OF THE LIVER
The liver has essential synthetic and excretory functions and can be thought of as a large ‘metabolic factory’. It also detoxifies and, like the kidneys, excretes the end products of metabolism. The main blood supply to the liver is via the portal vein. The liver is made up of hexagonal lobules of cells (Fig. 17.1). Rows of hepatocytes radiate from the central hepatic vein and are separated by sinusoidal spaces, along the walls of which are interspersed hepatic macrophages, the Kupffer cells. These phagocytic cells are part of the reticuloendothelial system and have an important detoxifying function. At the corners of each lobule are the portal tracts that contain branches of the hepatic artery, the portal vein and bile ducts. Blood flows from the portal tracts towards the central hepatic vein. Therefore:
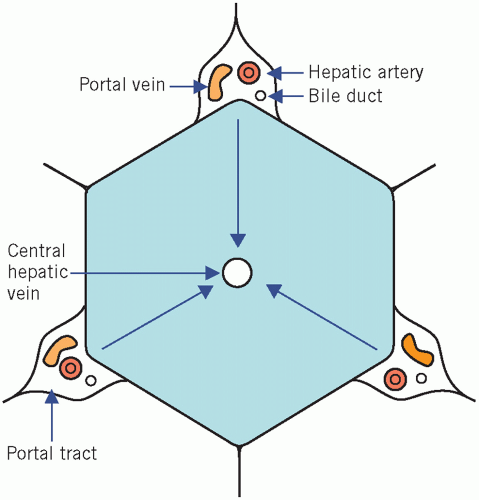 Figure 17.1 Diagrammatic representation of a crosssection of a hepatic lobule showing the relation between the central hepatic vein and the portal tracts. Blood flows towards the central vein, as indicated by the arrows. |
Hypoxia and toxins that are metabolized in the liver cause damage to the centrilobular area first.
Toxins that do not depend on hepatic metabolism primarily affect the periphery of the lobule.
Almost all nutrients from the gastrointestinal tract pass through the sinusoidal spaces prior to entering the systemic circulation. The hepatic architecture may be disturbed in cirrhosis (fibrosis).
General metabolic functions
When the glucose concentration is high in the portal vein, it is converted to glycogen and the carbon skeletons of fatty acids, which are transported to adipose tissue as very low-density lipoprotein (VLDL). During fasting, the systemic plasma glucose concentration is maintained by the breakdown of glycogen (glycogenolysis) or by the synthesis of glucose from substrates such as glycerol, lactate and amino acids (gluconeogenesis). Fatty acids reaching the liver from fat stores may be metabolized in the tricarboxylic acid cycle, converted to ketones or incorporated into triglycerides (see Chapter 13).
Synthetic functions
Hepatocytes synthesize:
plasma proteins, excluding immunoglobulins and complement,
most coagulation factors, including fibrinogen and factors II (prothrombin), V, VII, IX, X, XI, XII and XIII – of these, prothrombin (II) and factors VII, IX and X cannot be synthesized without vitamin K,
primary bile acids,
The liver has a very large functional reserve. Deficiencies in synthetic function can be detected only if liver disease is extensive. Before a fall in plasma albumin concentration is attributed to advanced liver disease, extrahepatic causes must be excluded, such as the loss of protein through the kidney, gut or skin, or across capillary membranes into the interstitial space, as in even mild inflammation or infection (see Chapter 19).
Prothrombin levels, assessed by measuring the prothrombin time, may be reduced because of impaired hepatic synthesis, whether due to failure to absorb vitamin K or to hepatocellular damage. If hepatocellular function is adequate, parenteral administration of vitamin K may reverse the abnormality.
Excretion and detoxification
The excretion of bilirubin is considered in more detail below. Other substances that are inactivated and excreted by the liver include the following:
Cholesterol – excreted in the bile either unchanged or after conversion to bile acids.
Amino acids – which are deaminated in the liver. Amino groups, and the ammonia produced by intestinal bacterial action and absorbed into the portal vein, are converted to urea.
Steroid hormones – which are metabolized and inactivated by conjugation with glucuronate and sulphate and excreted in the urine in these watersoluble forms.
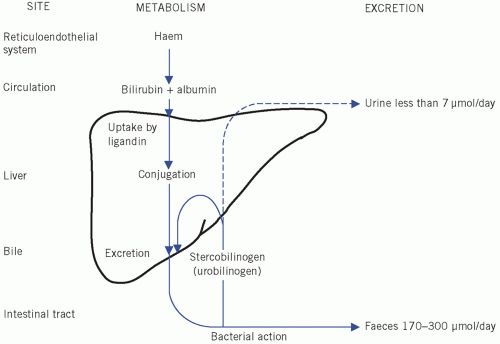
Figure 17.2 Metabolism and excretion of bilirubin.
Many drugs – which are metabolized and inactivated by enzymes of the endoplasmic reticulum system; some are excreted in the bile.
Toxins – the reticuloendothelial Kupffer cells in the hepatic sinusoids are well placed to extract toxic substances that have been absorbed from the gastrointestinal tract.
Efficient excretion of the end products of metabolism and of bilirubin depends on:
normally functioning liver cells,
normal blood flow through the liver,
patent biliary ducts.
Formation and excretion of bilirubin (Fig. 17.2)
At the end of their lifespan, red blood cells are broken down by the reticuloendothelial system, mainly in the spleen. The released haemoglobin is split into globin, which enters the general protein pool, and haem, which is converted to bilirubin after the removal of iron, which is reused (see Chapter 21).
About 80 per cent of bilirubin is derived from haem within the reticuloendothelial system. Other sources include the breakdown of immature red cells in the bone marrow and of compounds chemically related to haemoglobin, such as myoglobin and the cytochromes. Less than 300 µmol of bilirubin is produced daily from the breakdown of erythrocytes, while the normal liver is able to conjugate up to about 1 mmol/day, and
therefore hyperbilirubinaemia is an insensitive index of parenchymal hepatic disease.
therefore hyperbilirubinaemia is an insensitive index of parenchymal hepatic disease.
Bilirubin is normally transported to the liver bound to albumin. In this form it is called unconjugated bilirubin, which is lipid soluble and therefore, if not protein bound, can cross cell membranes, including those forming the blood-brain barrier. In this form it is potentially toxic; however, at physiological concentrations it is all protein bound.
In the adult, about 300 µmol per day of bilirubin reaches the liver, where it is transferred from plasma albumin, through the permeable vascular sinusoidal membrane. The hepatocytes can process a much greater load than this. Bilirubin is bound to ligandin (Y protein). From there it is actively transported to the smooth endoplasmic reticulum, where it is conjugated with glucuronate by a process catalysed by uridine diphosphate glucuronyl transferase.
Bilirubin monoglucuronide passes to the canalicular surfaces of the hepatocytes, where, after the addition of a second glucuronate molecule, it is secreted by active processes into the bile canaliculi. This process is largely dependent on the active secretion of bile acids from hepatocytes. These energy-dependent steps are the ones most likely to be impaired by liver damage (hypoxia and septicaemia) and by increased pressure in the biliary tract. Other anions, including drugs, may compete for binding to ligandin, thus impairing bilirubin conjugation and excretion. Novobiocin inhibits glucuronyl transferase, thus exacerbating unconjugated hyperbilirubinaemia. Bilirubin is often assayed by the Van den Bergh reaction, which allows conjugated (direct-reacting) and unconjugated (indirect-reacting) bilirubin to be distinguished.
Bilirubin metabolism and jaundice
Jaundice usually becomes clinically apparent when the plasma bilirubin concentration reaches about 50 µmol/L (hyperbilirubinaemia), about twice the upper reference limit. It occurs when bilirubin production exceeds the hepatic capacity to excrete it. This may be because:
An increased rate of bilirubin production exceeds normal excretory capacity of the liver (prehepatic jaundice).
The normal load of bilirubin cannot be conjugated and/or excreted by damaged liver cells (hepatic jaundice).
The biliary flow is obstructed, so that conjugated bilirubin cannot be excreted into the intestine and is regurgitated into the systemic circulation (posthepatic jaundice).
Retention of bilirubin in plasma: jaundice
Unconjugated hyperbilirubinaemia occurs if there is:
a marked increase in the bilirubin load as a result of haemolysis, or of the breakdown of large amounts of blood after haemorrhage into the gastrointestinal tract or, for example, under the skin due to extensive bruising; in cases of haemolysis, plasma bilirubin rarely exceeds 75µmol/L,
impaired binding of bilirubin to ligandin or impaired conjugation with glucuronate in the liver.
In some pathological conditions, plasma unconjugated bilirubin levels may increase so much that they exceed the protein-binding capacity. The lipid-soluble, unbound bilirubin damages brain cells (kernicterus). This is most likely to occur in newborn, particularly premature, infants in whom the hepatic conjugating mechanisms are immature. In addition, the proportion of unbound, unconjugated bilirubin, and therefore the risk of cerebral damage, increases if:
plasma albumin concentration is low,
unconjugated bilirubin is displaced from binding sites, for example by high levels of free fatty acids or drugs such as salicylates or sulphonamides.
Unconjugated bilirubin is normally all protein bound and is not water soluble and therefore cannot be excreted in the urine. Patients with unconjugated hyperbilirubinaemia do not have bilirubinuria (‘acholuric jaundice’) such as Gilbert’s syndrome. Conjugated bilirubinaemia is one of the earliest signs of impaired hepatic excretion. In most cases of jaundice in adults, both conjugated and unconjugated fractions of bilirubin are increased in plasma but conjugated bilirubin predominates. Conjugated bilirubin is water soluble and is less strongly protein bound than the unconjugated form, and therefore can be excreted in the urine. Bilirubinuria is always pathological. Dark urine may be an early sign of some forms of hepatobiliary disease.
Conjugated bilirubin enters the gut lumen in bile; it is broken down by bacteria in the distal ileum and the colon into a group of products known as stercobilinogen (faecal urobilinogen). Some is absorbed into the portal circulation, most of which is re-excreted in bile (enterohepatic circulation). A small amount enters the systemic circulation and is excreted in the urine as urobilinogen, which can be oxidized to a coloured pigment, urobilin.
Urobilinogen
Urobilinogen, unlike bilirubin, is often detectable in the urine of normal people by testing with commercial strip tests, particularly if the urine, and therefore the urobilinogen, is concentrated. Urinary urobilinogen concentration is increased in the following situations.
When haemolysis is very severe: large amounts of bilirubin enter the intestinal lumen and are converted to stercobilinogen. An increased amount of urobilinogen is formed and absorbed. If the hepatic capacity to re-secrete it is exceeded, it is passed in the urine.
When liver damage impairs re-excretion of normal amounts of urobilinogen into the bile.
The colourless, unabsorbed stercobilinogen is oxidized to stercobilin, a pigment that contributes to the brown colour of faeces. Pale stools may, therefore, suggest biliary obstruction associated with an absence of urinary urobilinogen.
BIOCHEMICAL TESTS FOR LIVER DISEASE
Several biochemical tests constitute what are called the ‘liver function tests’. Different tests can give different information about hepatic dysfunction.
Hepatocyte damage
Strictly speaking, changes in plasma enzyme activity generally indicate liver cell membrane damage rather than hepatic function capacity. Because these enzymes are also present in other tissues, changes in plasma activities may reflect damage to those tissues rather than to the liver (see Chapter 18).
Aminotransferases (alanine and aspartate)
A rise in plasma aminotransferase activities is a sensitive indicator of damage to cytoplasmic and/or mitochondrial membranes. Plasma enzyme activities rise when the membranes of only very few cells are damaged. Liver cells contain more aspartate aminotransferase (AST) than alanine aminotransferase (ALT), but ALT is confined to the cytoplasm, in which its concentration is higher than that of AST. Raised plasma transaminase concentrations are indicative of hepatocyte damage, but do not necessarily reveal its mechanism.
In inflammatory or infective conditions, such as viral hepatitis, the cytoplasmic membrane sustains the main damage; leakage of cytoplasmic contents causes a relatively greater increase in plasma ALT than AST activities.
In infiltrative disorders in which there is damage to both mitochondrial and cytoplasmic membranes, there is a proportionally greater increase in plasma AST than ALT activity.
The relative plasma activities of ALT and AST may help to indicate the type of cell damage. The former is more specific for hepatic disease; AST may be present in skeletal muscle and is more sensitive than ALT. A plasma AST:ALT ratio of > 2 is suggestive but not diagnostic of alcoholic liver disease and a ratio < 1 suggests chronic viral hepatitis or hepatic steatosis (see Chapter 18).
Hepatic synthetic function
The measurement of plasma albumin and prothrombin time may be used to assess function. The hepatic synthetic and secretory capacities are large; only severe and usually prolonged liver disease, for example cirrhosis, demonstrably impairs albumin and prothrombin synthesis.
Albumin
Hypoalbuminaemia is such a common finding in many severe illnesses that it is a less specific indicator of impaired synthetic capacity than a prolonged prothrombin time. A plasma albumin concentration below the lower reference limit may imply hepatic disease chronicity. However, there are many other causes of a low plasma albumin concentration that are not due to hepatic disease (see Chapter 19).
Prothrombin time
The prothrombin time may be prolonged by cholestasis: fat-soluble vitamin K cannot be absorbed normally if fat absorption is impaired due to intestinal bile salt deficiency. The abnormality is then corrected by parenteral administration of the vitamin. A prolonged prothrombin time may also result from severe impairment of synthetic ability if the liver cell mass is greatly reduced; in such cases it is not corrected by parenteral administration of vitamin K.
Hepatic excretory function
A high plasma conjugated bilirubin concentration indicates impaired hepatic excretory function but as this is also raised in hepatocellular disease it is not specific for cholestasis. This may be accompanied by a high plasma alkaline phosphatase (ALP) activity, as we will now see. Jaundice has been described above.
Other tests for liver disease
Alkaline phosphatase
Alkaline phosphatase is derived from a number of different tissues, including the liver, the osteoblasts in bone and the placenta. Plasma activities rise in cholestatic liver disease because ALP synthesis is increased and the enzyme within the biliary tract is regurgitated into plasma. A raised ALP concentration in the presence of a raised γ-glutamyl transferase (GGT) concentration implies that the ALP is of hepatic origin.
γ-Glutamyl transferase
γ-Glutamyl transferase is an enzyme derived from the endoplasmic reticulum of the cells of the hepatobiliary tract. As this reticulum proliferates, for example in response to the prolonged intake of alcohol and of drugs such as phenobarbital and phenytoin, synthesis of the enzyme is induced and plasma GGT activity increases. Therefore, raised plasma activities do not necessarily indicate hepatocellular damage, but may reflect enzyme induction or cholestasis.
Biochemical tests can be used to investigate hepatic disorders, the mechanisms underlying which can be divided into three main groups; these often coexist, but one usually predominates in any particular condition (Table 17.1).
Liver-cell damage is characterized by the release of enzymes (AST and ALT) from damaged hepatocytes. Plasma ALT and AST activities are increased.
Cholestasis is characterized by retention of conjugated bilirubin and of ALP, and by increased ALP synthesis at the sinusoidal surface. Plasma conjugated bilirubin levels and ALP activities are increased.
Table 17.1 Typical biochemical features of certain hepatic disorders
Plasma albumin
Bilirubin
Acute alcoholic hepatitis
–
↑
↑
↑
↑↑
Acute viral hepatitis
–
↑↑
↑↑
↑
↑↑
Chronic viral hepatitis
– or ↓
↑ or –
↑
↑ or –
↑
Cirrhosis
↓
↑ or –
↑
↑
↑
Gilbert’s syndrome
–
↑
–
–
–
Primary biliary cirrhosis
– or ↓
↑↑
↑
↑↑
↑↑
Tumour secondaries
–
↑ or –
↑
↑↑
↑↑
↑, raised; -, normal; ↓, reduced.
ALT, alanine aminotransferase; ALP, alkaline phosphatase; GGT, γ-glutamyl transferase.
Reduced mass of hepatocytes, if considerable, is characterized by a reduction in albumin and prothrombin synthesis. The plasma albumin concentration is reduced and the prothrombin time is prolonged.
Urine tests useful in suspected hepatic disease
Fresh urine analysis may confirm the presence of bilirubin (conjugated hyperbilirubinaemia) and urobilinogen
Urine Multistix include stabilized, diazotized 2,4-dichloraniline, which reacts with bilirubin to form azobilirubin. The test will detect about 3 µmol/L of bilirubin. Drugs, such as large doses of chlorpromazine, may give false-positive results.
Urine Multistix also include paradimethylaminobenzaldehyde, which reacts with urobilinogen. Urine Multistix do not react with porphobilinogen. This test will detect urobilinogen in urine from some normal individuals. False-positive results may occur after taking drugs such as para–aminosalicylic acid and some sulphonamides.
Bile acid measurement in obstetric cholestasis
Raised total plasma bile acid concentrations in the third trimester of pregnancy associated with pruritus are suggestive of obstetric cholestasis, which can lead to both maternal and fetal morbidity and mortality. Elevation of plasma ALT concentration may follow the increase in the concentration of plasma bile salts (see Chapter 10).
New hepatic function tests
Owing to the very large hepatic reserve, tests for impairment of metabolic (including synthetic and
secretory) function are relatively insensitive indicators of liver disease. There are a number of new tests that are being devised to improve the accuracy of the diagnosis of hepatic disorders. Tests of hepatocellular activity have been proposed, such as galactose elimination capacity, the aminopyrine breath test, indocyanine green clearance, and monoethylglycinexylidide (MEGX) production. All these tests are indirect measures of hepatic activity that rely on measuring compounds or their metabolites after they have been acted on by the liver. As yet, they do not have a place in routine clinical diagnosis.
secretory) function are relatively insensitive indicators of liver disease. There are a number of new tests that are being devised to improve the accuracy of the diagnosis of hepatic disorders. Tests of hepatocellular activity have been proposed, such as galactose elimination capacity, the aminopyrine breath test, indocyanine green clearance, and monoethylglycinexylidide (MEGX) production. All these tests are indirect measures of hepatic activity that rely on measuring compounds or their metabolites after they have been acted on by the liver. As yet, they do not have a place in routine clinical diagnosis.
DISEASES OF THE LIVER
Cholestasis
Cholestasis may be either:
intrahepatic, in which bile secretion from the hepatocytes into the canaliculi is impaired, due to:
viral hepatitis,
drugs such as chlorpromazine or toxins such as alcohol,
inflammation of the biliary tract (cholangitis),
autoimmune disease (primary biliary cirrhosis),
cystic fibrosis,
extrahepatic, due to obstruction to the flow of bile through the biliary tract by:
biliary stones,
inflammation of the biliary tract,
pressure on the tract from outside by malignant tissue, usually of the head of the pancreas,
biliary atresia (rare).
It is essential to distinguish between intrahepatic and extrahepatic causes of cholestasis, as surgery may be indicated for the latter but is usually contraindicated for intrahepatic lesions. The biochemical findings may be similar:
Bilirubin concentrations in plasma may be normal if only part of the biliary system is involved by intrahepatic lesions such as cholangitis, early primary biliary cirrhosis or primary or secondary tumours. The unaffected areas can secrete bilirubin.
Alkaline phosphatase activity is a sensitive test for cholestasis. Increased synthesis of ALP in the affected ducts increases the activity of this enzyme in plasma. If this is the only abnormal finding, it must be shown to be of hepatic origin before it is assumed to indicate liver disease.
Patients with prolonged and more widespread cholestasis may present with severe jaundice and pruritus due to the deposition of retained bile salts in the skin; the plasma bilirubin concentration may be more than 800 µmol/L. More rarely, there is bleeding due to malabsorption of vitamin K, with consequent prothrombin deficiency. Cholesterol retention may cause hypercholesterolaemia. Dark urine and pale stools suggest biliary retention of conjugated bilirubin.
The jaundice caused by extrahepatic obstruction due to malignant tissue is typically painless and progressive, but there may be a history of vague persistent back pain and weight loss. By contrast, intraluminal obstruction by a gallstone may cause severe pain, which, like the jaundice, is often intermittent. Gallstones may not always cause such symptoms. If a large stone lodges in the lower end of the common bile duct, the picture may be indistinguishable from that of malignant obstruction.
Although most of the findings are directly attributable to cholestasis, biliary back pressure may damage hepatocytes, and plasma aminotransferase activities may increase. Unless the cause is clinically obvious, evidence of dilated ducts due to extrahepatic obstruction should be sought using tests such as ultrasound, computerized tomography (CT) scanning or cholangiography.
Primary biliary cirrhosis
This is a rare autoimmune disorder that occurs most commonly in middle-aged women. Destruction and proliferation of the bile ducts produce a predominantly cholestatic picture, with pruritus and a plasma ALP activity that may be very high. Jaundice develops late in most patients. Mitochondrial antibodies are detectable in the plasma of more than 90 per cent of cases; the plasma immmunoglobulin M (IgM) concentration is usually raised. Patients may also manifest hypercholesterolaemia, xanthelasma (see Chapter 13), other autoimmune disorders and osteoporosis.
Parenteral nutrition
Prolonged parenteral nutrition may be associated with a progressive increase in plasma ALP activity and a subsequent rise in the plasma aminotransferase concentrations. The cause is not known, although biliary sludging may occur and the biochemical changes may return to normal when the parenteral feeding is discontinued. Cyclical parenteral nutrition may help the situation when patients are fed intermittently (see Chapter 14).
CASE 1
A 52-year-old woman was referred to the hepatology clinic because of jaundice, pruritus, hepatomegaly, xanthelasma and the following abnormal liver test results:
Plasma
Bilirubin 93 µmol/L (< 20)
Alanine aminotransferase 111 U/L (< 42)
Alkaline phosphatase (ALP) 826 U/L (< 250)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


