Introduction
Although many different pathogenic agents and processes can affect the liver (Table 14–1), they are generally manifested in individual patients in a limited number of ways that can be assessed by evaluation of some key parameters. Liver disease can be acute or chronic, focal or diffuse, mild or severe, and reversible or irreversible. Most cases of acute liver disease (eg, caused by viral hepatitis) are so mild that they never come to medical attention. Transient symptoms of fatigue, loss of appetite, and nausea are often ascribed to other causes (eg, flu), and minor biochemical abnormalities referable to the liver that would be identified in blood studies are not discovered. The patient recovers without any lasting medical consequences. In other cases of acute liver injury, symptoms and signs are severe enough to call for medical attention. The entire range of liver functions may be affected or only a few, as is the case with liver injury resulting from certain drugs that cause isolated impairment of the liver’s role in bile formation (cholestasis). Occasionally, viral, drug-induced, and other acute liver injury occurs in an overwhelming manner, resulting in massive liver cell death and progressive multiorgan failure. This syndrome of acute liver failure (also referred to as fulminant hepatic failure) carries a high mortality rate; however, in recent years, use of emergency liver transplantation has significantly improved survival.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Liver injury may continue beyond the initial acute episode or may be recurrent (chronic hepatitis). In some cases of chronic hepatitis, liver function remains stable or the disease process ultimately resolves altogether. In other cases, there is progressive and irreversible deterioration of liver function.
Cirrhosis is ultimately the consequence of progressive liver injury. Cirrhosis can occur in a subset of cases of chronic hepatitis that do not resolve spontaneously or after repeated episodes of acute liver injury, as in the case of chronic alcoholism. In cirrhosis, the liver becomes hard, shrunken, and nodular and displays impaired function and diminished reserve because of a decreased amount of functioning liver tissue. More importantly, the physics of blood flow is altered such that the pressure in the portal vein is elevated. As a result, the blood is diverted around the liver rather than filtered through the liver. This phenomenon, termed portal-to-systemic (or portosystemic) shunting, has profound effects on the function of various organ systems and sets the stage for certain devastating complications of liver disease that are described later.
Although liver disease resulting from many different causes may present in common ways, the reverse is also true (ie, liver disease from specific causes may have distinctly different presentations in different patients). For example, consider two patients with acute viral hepatitis: One may present with yellow eyes and skin—a manifestation of impaired liver function—complaining of nothing more than itching, fatigue, and loss of appetite, whereas the other may be brought to the emergency room moribund, with massive gastrointestinal (GI) bleeding and encephalopathy. Such variations in the severity of liver disease are probably due to genetic, immunologic, and environmental (including perhaps nutritional) factors that are currently poorly understood.
The consequences of liver disease can be either reversible or irreversible. Those arising directly from acute damage to the functional cells of the liver, most notably hepatocytes, without destruction of the liver’s capacity for regeneration, are generally reversible. Like many organs of the body, the liver normally has both a huge reserve capacity for the various biochemical reactions it carries out and the ability to regenerate fully differentiated cells and thereby recover completely from acute injury. Thus, only in the most fulminant cases or in end-stage disease are there insufficient residual hepatocytes to maintain minimal essential liver functions. More commonly, patients display transient signs of liver cell necrosis and disordered function followed by full recovery. The symptoms and signs of this sort of acute liver injury can best be understood as an impairment of normal biochemical functions of the liver.
Other consequences of liver disease are irreversible, typically seen in the patient with cirrhosis. These are best understood as a result of portosystemic shunting of blood flow. They include a heightened sensitivity to noxious substances absorbed from the GI tract (encephalopathy), an increased risk of massive GI bleeding (development of varices and coagulopathy), and malabsorption of fat in the stool (as a result of decreased bile flow). Nevertheless, some of these consequences are treatable. Commonly, patients with cirrhosis present with superimposed acute liver injury (eg, caused by an alcoholic binge or other drug exposure). Because they have a decreased hepatocyte mass and much less functional reserve, they are more sensitive to acute liver injury than patients with a normal liver.
Checkpoint
What parameters must you consider in assessing a patient with liver disease?
What factors may determine the difference in severity of liver disease between two patients with acute hepatitis resulting from the same cause?
In what ways is the patient with underlying cirrhosis who presents with acute hepatitis likely to be different from the patient with a previously normal liver and acute hepatitis?
Structure & Function of the Liver
The liver is located in the right upper quadrant of the abdomen in the peritoneal space just below the right side of the diaphragm and under the rib cage (Figure 14–1). It is anatomically separated into two predominant lobes, a right and a left lobe. The right lobe has two lesser segments, the posterior caudate lobe and the inferior quadrate lobe. The liver can also be differentiated functionally via the portal blood flow into four sectors, which are further subdivided into eight segments. The liver weighs approximately 1400 g in the adult and is covered by a fibrous capsule. It receives nearly 25% of the cardiac output, approximately 1500 mL of blood flow per minute, via two sources: venous flow from the portal vein, which is crucial to performance of the liver’s roles in bodily functions, and arterial flow from the hepatic artery, which is important for liver oxygenation and which supplies the biliary system via the cystic artery. These vessels converge within the liver, and the combined blood flow exits via the so-called central veins (also called terminal veins or hepatic venules) that drain into the hepatic vein and ultimately the inferior vena cava.
The portal vein carries venous blood from the small intestine, rich in freshly absorbed nutrients—as well as drugs and poisons—directly to the liver. Also flowing into the portal vein before its entry into the liver is the pancreatic venous drainage, rich in pancreatic hormones (insulin, glucagon, somatostatin, and pancreatic polypeptide). The portal vein forms a specialized capillary bed that allows individual hepatocytes to be bathed directly in portal blood. In part because of this system of blood supply, the liver is a prime site for metastatic spread of cancer, especially from the GI tract, breast, and lung.
The substance (parenchyma) of the liver is organized into plates of hepatocytes lying in a cage of supporting cells termed reticuloendothelial cells (Figure 14–2A). The plates of hepatocytes are generally only one cell thick, and individual plates are separated from each other by vascular spaces called sinusoids. It is in these sinusoids that blood from the hepatic artery is mixed with blood from the portal vein on the way to the central vein. The reticuloendothelial cell meshwork in which the hepatocytes reside includes diverse cell types, most importantly the endothelial cells that make up the walls of the sinusoids; specialized macrophages, termed Kupffer cells, which are anchored in the sinusoidal space; and stellate cells or lipocytes, fat-storing cells involved in vitamin A metabolism, which lie between the hepatocytes and the endothelial cells. Approximately 30% of all cells in the liver are reticuloendothelial cells, and about 33% of these are Kupffer cells. Yet, because reticuloendothelial cells are smaller than hepatocytes, the reticuloendothelial system accounts for only 2–10% of the total protein in the liver. The reticuloendothelial cells are much more than just a cage for hepatocytes. They perform specific functions, including phagocytosis and secretion of cytokines, and communicate with each other as well as with hepatocytes. Their dysfunction contributes to both hepatocyte necrosis in acute liver disease and to hepatic fibrosis in chronic liver disease.
Figure 14–2
A: Detailed structure of the liver lobule. (Redrawn, with permission, from Chandrasoma P et al. Concise Pathology, 3rd ed. Originally published by Appleton & Lange. Copyright © 1998 by The McGraw-Hill Companies, Inc.) B: Relationship of lobule to acinus. (CV, central vein; PS, portal space [or triad].) (Redrawn, with permission, from Leeson CR. Histology, 2nd ed. Saunders, 1970.) C: Hepatic acinus. (HV, hepatic venule.) (Reproduced, with permission, from Gumucio JJ. Hepatic transport. In: Kelley WN (ed): Textbook of Medicine. Lippincott, 1989.)
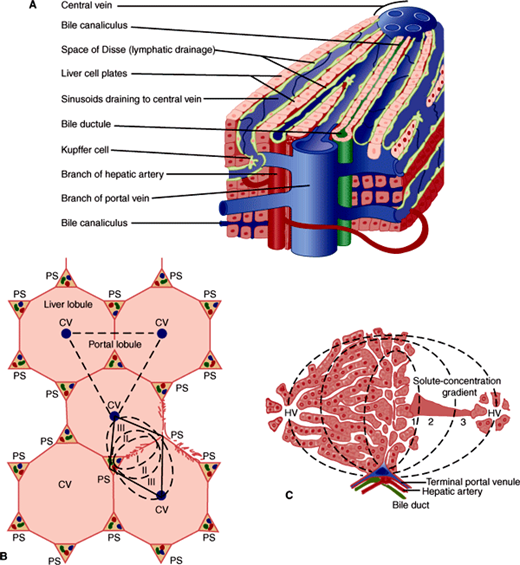
Under the microscope at low-power magnification, liver architecture has been traditionally described in terms of the lobule (Figure 14–2B). Neat arrays of hepatocyte plates are organized around individual central veins to form hexagons with portal triads or tracts (sheathlike structures containing a portal venule, hepatic arteriole, and bile canaliculus) at their corners. The hepatocytes adjacent to the portal triad are termed the limiting plate. Disruption of the limiting plate is a significant diagnostic marker of some forms of immune-mediated liver disease. This may be seen in liver biopsies from patients with liver disease of unknown cause.
Physiologically, it is more useful to think of liver architecture in terms of the portal-to-central direction of blood flow: Blood entering the sinusoids from a terminal portal venule or hepatic arteriole flows past hepatocytes closest to those vessels first (termed zone 1 hepatocytes) and then percolates past zone 2 hepatocytes (so called because they are not the first hepatocytes reached by blood entering the hepatic parenchyma). The last hepatocytes reached by the blood before it enters the central vein are termed zone 3 hepatocytes. Thus, the microscopic organization of the liver can be viewed in terms of functional zones. A liver acinus is defined as the unit of liver tissue centered around the portal venule and hepatic arteriole whose hepatocytes can be imagined to form concentric rings of cells in the order in which they come into contact with portal blood, first to last (Figure 14–2C). Hepatocytes at either extreme of the acinus (zones 1 and 3) appear to differ in both enzymatic activity and physiologic functions. Zone 1 hepatocytes, exposed to the highest oxygen concentrations, are particularly active in gluconeogenesis and oxidative energy metabolism. They are also the major site of urea synthesis (because freely diffusible substances such as ammonia absorbed from protein breakdown in the gut are largely extracted in zone 1). Conversely, zone 3 hepatocytes are more active in glycolysis and lipogenesis (processes requiring less oxygen). Zone 2 hepatocytes display attributes of both zone 1 and zone 3 cells.
Functional zonation applies only to processes driven by the presence of diffusible substances. The liver, however, is also involved in many pathways participating in receptor-mediated uptake and active transport of substances that are unable to diffuse freely into cells. These substances enter whichever hepatocytes have the appropriate transporters regardless of their zone. Similarly, substances that are tightly bound to carrier proteins for which the liver does not have receptors are cleared equally poorly by hepatocytes in all three zones.
All surfaces of a hepatocyte are not the same. Hepatocytes have three sides. One side, the apical surface, forms the wall of the bile canaliculus. The second side, the basolateral surface, is in contact with the bloodstream via the sinusoids. The last side, the lateral domain, is bordered by the two other surfaces. Very different activities go forward at these regions of the hepatocyte plasma membrane; tight junctions between hepatocytes serve to maintain segregation of apical and basolateral plasma membrane domains. Processes related to bile transport and excretion act at the apical plasma membrane (Figure 14–3A). Uptake from and secretion into the bloodstream are activities that occur across the basolateral membrane (Figure 14–3B).
Figure 14–3
A: Mechanism of secretion of bile acids. About 90% of these compounds derive from bile acids absorbed in the intestinal epithelium and recirculated to the liver. The remainder are synthesized in the liver by conjugating cholic acid with the amino acids glycine and taurine. This process occurs in the smooth endoplasmic reticulum (SER). B: Protein synthesis and carbohydrate storage in the liver. Protein synthesis occurs in the rough endoplasmic reticulum, which explains why liver cell lesions or starvation leads to a decrease in the amounts of albumin, fibrinogen, and prothrombin in a patient’s blood. In several diseases, glycogen degradation is depressed, with abnormal intracellular accumulation of this compound. (SER, smooth endoplasmic reticulum; RER, rough endoplasmic reticulum.) (Redrawn, with permission, from Junqueira LC et al. Basic Histology, 10th ed. McGraw-Hill, 2003.)
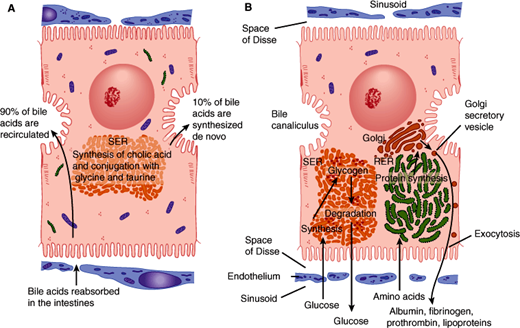
In view of this organization, it is perhaps not surprising that hepatocyte dysfunction can sometimes involve disruption of bile flow (cholestasis) with relative preservation of other functions. There is, however, no clear line between the consequences of disturbed apical and basolateral functions: Cholestasis, although initially a disorder of apical bile flow, is ultimately manifested at the basolateral surface. This is because it is at the basolateral surface that bilirubin and other substances to be excreted across the apical plasma membrane into the bile must first be taken up from the bloodstream. Similarly, disruption of energy metabolism or protein synthesis, although initially impinging on the secretory and metabolic processes of the hepatocyte, will ultimately affect the bile transport machinery in the apical plasma membrane as well.
Although the normal liver contains very few cells in mitosis, when hepatocytes are lost, poorly understood mechanisms stimulate proliferation of the remaining hepatocytes. This is why in most cases of fulminant hepatic failure with massive hepatocellular death, if the patient survives the acute period of hepatic dysfunction (usually with medical therapy in the hospital), recovery will be complete. Similarly, surgical resection of liver tissue is followed by proliferation of the remaining hepatocytes (hyperplasia). Numerous growth factors (eg, HGF, TGF-α) and cytokines (eg, TNF, IL-1, IL-6) are involved in positioning the liver on a continuum between cell proliferation and cell death.
Checkpoint
From which vascular beds do the hepatic central veins derive their blood flow?
Why is the liver a major site for metastasis of malignant neoplasms from other parts of the body?
What cell types make up the liver, and what are their distinguishing characteristics?
What is the difference between the lobule concept and the acinus concept of liver subarchitecture?
What are some physiologic consequences of functional zonation in the liver?
What activities are found in zone 1 hepatocytes? In zone 3 hepatocytes?
What structures normally maintain the separation of apical and basolateral plasma membrane domains of the hepatocyte?
What happens to the remaining hepatocytes when part of the liver is surgically resected?
The portal blood flow, being venous in nature, is normally under low hydrostatic pressure (about 10 mm Hg). Accordingly, there must be little resistance to its flow within the liver, allowing the blood to percolate through the sinusoids and achieve maximal contact—for exchange of substances—with hepatocytes. Two unique features—fenestrations in the endothelial cells and lack of a typical basement membrane between endothelial cells and hepatocytes—aid in making the liver a low-pressure circuit for the flow of portal blood. These features are altered in cirrhosis, resulting in increased portal pressure and profound changes in liver blood flow, with devastating clinical consequences.
Fenestrations are spaces between the endothelial cells that make up the walls of the portal capillary system, which allow plasma and its proteins, but not red blood cells, free and direct access to the surface of the hepatocytes. This feature is crucial to the liver’s function of uptake from and secretion into the bloodstream. This feature also contributes to the efficiency of the liver as a filter of portal blood. Most of the capillary beds in the body lack such fenestrations.
The diverse functions of the liver are listed as four broad categories in Table 14–2. Although there is considerable overlap between them, systematic consideration of each category is a useful way of approaching the patient with liver disease.
|
|
|
|
|
|
|
|
Much of the body’s carbohydrate, lipid, and protein is synthesized, metabolized, and interconverted in the liver; products are removed from or released into the bloodstream in response to the energy and substrate needs of the body.
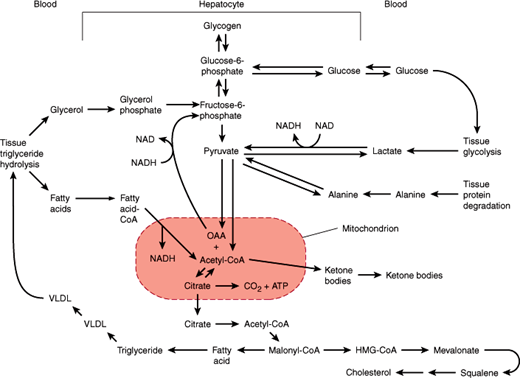
After a meal, the liver achieves net glucose consumption (eg, for glycogen synthesis and generation of metabolic intermediates via glycolysis and the tricarboxylic acid cycle). This occurs as a result of a confluence of several effects. First, the levels of substrates such as glucose increase. Second, the levels of hormones that affect the amount and activity of metabolic enzymes change. Thus, when blood glucose levels increase, the ratio of insulin to glucagon in the bloodstream also increases. The net effect is increased glucose utilization by the liver. In times of fasting (low blood glucose) or stress (when higher blood glucose is needed), hormone and substrate levels in the bloodstream drive metabolic pathways of the liver responsible for net glucose production (eg, the pathways of glycogenolysis and gluconeogenesis). As a result, blood glucose levels are raised to, or maintained in, the normal range in spite of wide and sudden changes in the rate of glucose input (eg, ingestion and absorption) and output (eg, utilization by tissues) from the bloodstream (Figure 14–4).
Figure 14–4
Pathways of hepatic carbohydrate and lipid metabolism. (ATP, adenosine triphosphate; CoA, co-enzyme A; HMG, hepatic 3-methylglutaryl; OAA, oxaloacetic acid; VLDL, very low-density lipoproteins.) (Redrawn from Schwartz CC. Hepatic metabolism. In: Kelley WN, ed. Textbook of Medicine. Lippincott, 1989.)
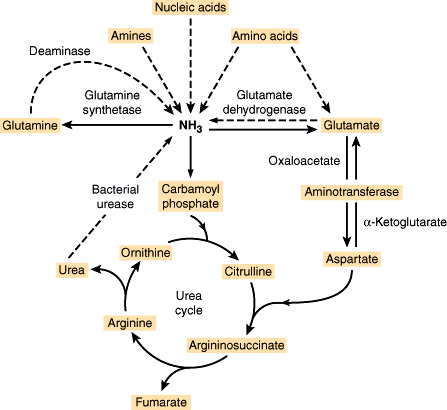
Related to its important role in protein metabolism, the liver is a major site for processes of oxidative deamination and transamination (Figure 14–5). These reactions allow amino groups to be shuffled among molecules in order to generate substrates for both carbohydrate metabolism and amino acid synthesis. Likewise, the urea cycle allows nitrogen to be excreted in the form of urea, which is much less toxic than free amino groups in the form of ammonium ions. Impairment of this function in liver disease is discussed in greater detail later.
Figure 14–5
Urea cycle. Dashed lines signify pathways whose extent of involvement varies from patient to patient depending on genetic, dietary, and other factors. (Redrawn, with permission, from Powers-Lee SG et al. Urea synthesis and ammonia metabolism. In: Arias IM et al, eds. The Liver: Biology and Pathology, 3rd ed. Raven Press, 1994.)
The liver is the center of lipid metabolism. It manufactures nearly 80% of the cholesterol synthesized in the body from acetyl-CoA via a pathway that connects metabolism of carbohydrates with that of lipids (Figure 14–4). Moreover, the liver can synthesize, store, and export triglycerides (Figure 14–4). The liver is also the site of keto acid production via the pathway of fatty acid oxidation that connects lipid catabolism with activity of the tricarboxylic acid cycle.
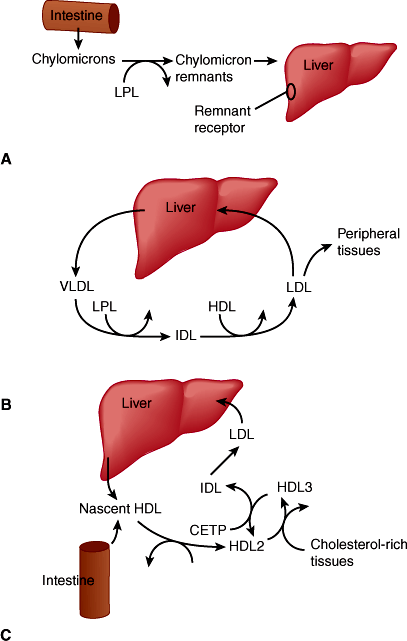
In the process of controlling the body’s level of cholesterol and triglycerides, the liver assembles, secretes, and takes up various lipoprotein particles (Figure 14–6). Dietary fat is first absorbed into the small intestine then packaged into chylomicrons. Following removal of triglycerides, the chylomicron remnant is taken up by the liver via low-density lipoprotein (LDL) receptor–mediated endocytosis. To distribute lipids systemically, very-low-density lipoproteins (VLDLs) are secreted by the liver and transport triglycerides and cholesterol to adipose tissue for storage or to other tissues for immediate use. As triglycerides are removed, the structure of VLDL particles is modified by loss of lipid and protein components rendering intermediate-density lipoprotein (IDL) and further downstream LDL. LDL particles are then returned to the liver via the LDL receptor. On the other hand, high-density lipoproteins (HDLs), a lipoprotein synthesized and secreted from the liver, scavenge excess cholesterol and triglycerides from other tissues and from the bloodstream, returning them to the liver where they are excreted. Thus, secretion of HDL and removal of LDL are both mechanisms by which cholesterol in excess of that needed by various tissues is removed from the circulation (Figures 14–6B and 14–6C).
Figure 14–6
Lipoprotein metabolism in the liver. A: Exogenous fat transport pathway. B: Endogenous fat transport pathway. C: Pathway of reverse cholesterol transport. In each of these three pathways, lipoprotein particles are used to solubilize cholesteryl esters (and triglyceride), either for the purpose of import from the GI tract (A), distribution to various tissues (B), or transport to the liver for excretion in bile (C). During their circulation, specific lipoprotein particles are transformed by addition and removal of apoproteins and by the action of enzymes in plasma or in tissues (eg, LPL, lipoprotein lipase; CETP, cholesteryl ester transfer protein). Intermediate-density lipoproteins (IDL) are intermediates in the conversion of VLDL to LDL. (HDL, high-density lipoproteins; LDL, low-density lipoproteins.) (Redrawn from Breslow JL. Genetic basis of lipoprotein disorders. J Clin Invest. 1989;84:373.)
The liver manufactures and secretes many of the proteins found in plasma, including albumin, several of the clotting factors, a number of binding proteins, and even certain hormones and hormone precursors. By virtue of the actions of these proteins, the liver has important roles in maintaining plasma oncotic pressure (serum albumin), coagulation (clotting factor synthesis and modification), blood pressure (angiotensinogen), growth (insulin-like growth factor-1), and metabolism (steroid and thyroid hormone–binding proteins).
The liver plays an important role in solubilizing, transporting, and storing a variety of very different substances that would otherwise be difficult for the tissues to obtain or to move in and out of cells. Specific cells in the liver perform these functions by manufacturing specialized proteins that serve as receptors, binding proteins, or enzymes.
Bile is a detergent-like substance synthesized by the liver that permits a variety of otherwise insoluble substances to be dissolved in an aqueous environment for transport into or out of the body. Bile acids are a major component of bile and are recycled via the so-called enterohepatic circulation between the liver and the intestines. After synthesis and active transport from hepatocyte cytoplasm into the bile canaliculus (across the apical plasma membrane of the hepatocyte), bile is collected in the biliary tract (and sometimes stored in the gallbladder) and excreted via the common bile duct into the duodenum. While still in the cytoplasm of the hepatocyte, many bile acids are conjugated to sugars, which increases their water solubility. Once in the duodenum, bile acids serve to solubilize lipids, facilitating digestion and absorption of fats. In the terminal ileum, both conjugated and deconjugated bile acids are taken up and transported from enterocytes to portal blood flow. Portal blood returns them to the liver, where specialized bile acid transporters (predominantly sodium taurocholate cotransporter, or Ntcp) return them to the hepatocyte cytosol via the basolateral plasma membrane facing the space of Disse. There they are subject to reconjugation and secretion across the apical membrane along with other components (eg, pigment, cholesterol) to form new bile. Thereafter, they engage in another cycle of enterohepatic transport.
Most of the enzymes that catalyze metabolic processes necessary for the detoxification and excretion of drugs and other substances are located in the smooth endoplasmic reticulum of hepatocytes. These pathways are used not only for metabolism of exogenous drugs but also for many endogenous substances that would otherwise be difficult for cells to excrete (eg, bilirubin and cholesterol). In most cases, this metabolism involves the conversion of hydrophobic (lipophilic) substances (which are difficult to excrete from cells because they tend to partition into cellular membranes) into more hydrophilic (polar) substances. This process involves catalysis of covalent modifications to make the substance more charged, so that it will partition more readily into an aqueous medium or at least be solubilized sufficiently in bile. As a result of these processes, collectively termed biotransformations, some substances that would otherwise be retained in cellular membranes can be excreted directly in the urine or transported into the bile for excretion in feces.
Biotransformation generally occurs in two phases. Phase I reactions involve oxidation reductions in which an oxygen-containing functional group is added to the substance to be excreted. While oxidation itself does not necessarily have a major effect on water solubility, it usually introduces into the drug a reactive “handle” that makes possible other reactions that do render the modified substance water soluble. These phase II reactions usually involve covalent attachment of the drug to a water-soluble carrier molecule such as the sugar glucuronic acid or the peptide glutathione. Unfortunately, by making substances more chemically reactive, phase I oxidation reactions often convert mildly toxic drugs into more toxic reactive intermediates. If conjugation by phase II enzymes is impaired for some other reason, the reactive intermediate can sometimes react with and damage other cellular structures. This feature of drug detoxification has important clinical implications.
The detoxification and bile transport pathways allow hepatocytes to convert a wide range of hydrophobic low- molecular-weight substances (eg, drugs and bilirubin) into a more hydrophilic and hence water-soluble form in which they can be excreted (eg, in bile or by the kidney). However, these are not the only solubilization challenges facing the body. The body also needs a mechanism that makes lipids available to various tissues (eg, to synthesize membranes) and one that removes any excess lipid the tissues do not use. For these processes to occur, lipid must be solubilized in a dispersed form that can be carried through the bloodstream. For this purpose, hepatocytes synthesize a class of specialized apolipoproteins. Apolipoproteins assemble into a variety of lipoprotein particles that transport lipids to and from various tissues by receptor-mediated endocytosis (see prior discussion of lipid metabolism).
Various cells in the liver synthesize proteins that bind certain substances very tightly (eg, some vitamins, minerals, and hormones). In some cases, this allows their transport in the bloodstream, where they would otherwise not be soluble (eg, steroids bound to steroid-binding globulin, which is synthesized and secreted by hepatocytes). In other cases, binding proteins made by the liver (eg, thyroid hormone–binding globulin) allow transport of specific substances (eg, thyroxine) in a form not fully accessible to tissues. In this way, the effective concentration of the substance is limited to its free concentration at equilibrium, and the tightly bound fraction forms a reservoir of the substance that is made available slowly as the free fraction is metabolized, thereby prolonging its half-life.
In some cases, binding proteins allow the liver to accumulate specific substances in relatively high concentrations and store them in a nontoxic form. Consider iron, for example, an essential nutrient. Free iron can be quite toxic to cells both directly as an oxidant and indirectly as an essential nutrient needed by infectious agents. Control of body iron occurs at the level of the enterocyte in the duodenum (see Chapter 13). Thus, the primary defect in the iron overload disorder hemochromatosis probably involves the enterocyte. Nevertheless, the liver has the responsibility of making a variety of proteins crucial for the binding and metabolism of iron. Through the actions of these proteins, the body gets the iron it needs without allowing excess free iron to cause damage or support pathogens.
Transferrin is an iron-binding protein synthesized and secreted into the bloodstream by the liver. On binding of free iron at normal pH, transferrin undergoes a conformational change that gives it high affinity for a specific membrane receptor of the hepatocyte (transferrin receptor). On receptor binding, the transferrin–transferrin receptor complex is internalized into the endocytic pathway, a progressively more acidic environment. There, at low pH, iron no longer remains bound to transferrin. However, conformational changes that occur at low pH allow transferrin to maintain high-affinity binding to its receptor even in the absence of bound iron. Thus, when the receptor recycles back to the surface, it brings the “empty” transferrin with it. On presentation to the pH 7.4 environment of the bloodstream, transferrin lacking bound iron is released from the receptor, and the cycle can start over again. In this way, transferrin and its receptor keep the bloodstream free of unbound iron. Meanwhile, the free iron released from transferrin in the acidic environment of the endosome is transported into the cytoplasm of the hepatocyte, where it binds to ferritin, a cytoplasmic iron storage protein. This provides a reservoir that can be mobilized in response to the body’s needs but makes iron inaccessible to pathogens and keeps it from causing direct toxic effects. Similar dynamics of plasma-binding proteins, receptors, or cytosolic storage proteins occur for many other substances, including fat-soluble vitamins and steroid hormones.
Whereas most solubilization functions are performed in hepatocytes, some of the binding and storage functions involve accessory cells. Thus, vitamin A storage occurs in fat droplets seen in the lipocytes of the reticuloendothelial system. Lipocytes have been implicated in the pathogenesis of chronic liver injury and cirrhosis. Injury to other cells releases cytokines, which activate the lipocytes. The lipocytes respond by proliferating and by synthesizing collagen and other basement membrane components, leading to an increase in the extracellular matrix and contributing to hepatic fibrosis.
Many of the functions of the liver already discussed (eg, drug detoxification and excretion of excess cholesterol by conversion to and solubilization in bile) can also be considered protective. Nevertheless, it is useful to conceptualize the protective function as a separate category because of its clinical importance in ameliorating the consequences of liver disease.
The liver helps remove bacteria and antigens that breach the defenses of the gut to enter the portal blood and also participates in clearing the circulation of endogenously generated cellular debris. It appears that specialized receptors on the Kupffer cell surface bind to glycoproteins (via carbohydrate receptors), to material coated with immunoglobulin (via the Fc receptor), or to complement (via the C3 receptor), thus allowing damaged plasma proteins, activated clotting factors, immune complexes, senescent blood cells, and so on to be recognized and removed.
Hepatocytes have a number of specific receptors for damaged plasma proteins distinct from the receptors present on Kupffer cells (eg, the asialoglycoprotein receptor that specifically binds glycoproteins whose terminal sialic acid sugar residues have been removed). The precise physiologic significance of this metabolic action remains unclear.
Ammonia generated from deamination of amino acids is metabolized within hepatocytes into the much less toxic substance urea. Loss of this function results in altered mental status, a common manifestation of severe or end-stage liver disease.
Glutathione is the major intracellular (cytoplasmic) reducing reagent and thus is crucial for preventing oxidative damage to cellular proteins. This molecule is a nonribosomally synthesized tripeptide (γ-glutamyl-cystinyl-glycine) that is also a substrate for many phase II drug detoxification conjugation reactions. The liver may also export glutathione for use by other tissues.
Some additional indirect liver functions (eg, its role in maintaining normal sodium and water balance) are inferred from the derangements observed in patients with liver disease, as discussed in the following section.
A number of blood tests are commonly used to assess liver injury. Serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) are measurements of levels of enzymes normally situated within hepatocytes. Their presence in the serum is thus actually a sign of liver cell necrosis rather than an indication of liver function.
To assess liver function more directly, a number of other tests can be used. The levels of albumin, clotting factors, and bilirubin can be measured in blood samples. Each of these tests has advantages and disadvantages, and no one of them serves as an ideal sole indicator of liver function. For example, albumin has a relatively long half-life (18–20 days); its synthesis can be stimulated in excess of need, and it can be lost via the kidneys in renal disease. Furthermore, about two thirds of body albumin is located in the extravascular, extracellular space, so changes in fluid distribution can alter serum albumin concentration. Likewise, the simplest measure of clotting factor levels, the prothrombin time (PT), is a relatively insensitive measure because it does not become abnormal until more than 80% of hepatic synthetic capacity is lost. Furthermore, vitamin K deficiency, occurring in patients with nutritional deprivation, chronic cholestasis, or fat malabsorption, can prolong the PT. Serum bilirubin is a good measure of cholestasis, and determination of conjugated (direct) versus unconjugated (indirect) bilirubin provides a good assessment of whether cholestasis is intrinsic to the liver or due solely to obstruction (eg, by a stone in the common bile duct). Furthermore, cholestasis, even when caused by liver disease, often does not reflect the degree to which other hepatic functions are lost, and unconjugated hyperbilirubinemia can occur for other reasons (eg, hemolysis).
For these reasons, an accurate assessment of liver function requires several blood tests (eg, AST, ALT, albumin, PT, bilirubin) as well as clinical assessment of the patient.
The two most common schemes for grading liver function is the modified Child-Turcotte-Pugh (CTP) score (Table 14–3) and the Model for End-Stage Liver Disease (MELD score = 3.78[Ln serum bilirubin (mg/dL)] + 11.2[Ln INR] + 9.57[Ln serum creatinine (mg/dL)] + 6.43). The CTP score predicts 1- and 2-year survival, and the MELD score predicts short-term (3-month) survival. In the United States, the MELD score is currently used to prioritize patients for donor allocation and liver transplantation.
| Points | |||
| Parameter | 1 | 2 | 3 |
| Albumin | >3.5 g/dL | 2.8–3.5 g/dL | <2.8 g/dL |
| Bilirubin | <2.0 mg/dL | 2.0–3.0 mg/dL | >3.0 mg/dL |
| Prothrombin time prolongation | <4.0 s | 4.0–6.0 s | >6.0 s |
| Ascites | Absent | Controlled | Refractory |
| Encephalopathy | None | Controlled | Refractory |
Checkpoint
What are the roles of the liver in carbohydrate, protein, and lipid metabolism?
What are two physiologic mechanisms by which the body transports cholesterol?
Explain phase I and phase II reactions in drug detoxification.
Name and explain four clearance or protective functions of the liver.
What specializations allow the liver normally to be a low-pressure conduit for blood flow?
Overview of Liver Disease
Most of the clinical consequences of liver disease can be understood either as a failure of one of the liver’s four broad functions (summarized in Table 14–2) or as a consequence of portal hypertension, the altered hepatic blood flow of cirrhosis.
One mechanism of liver disease, particularly in acute liver injury, is dysfunction of the individual hepatocytes that make up the liver parenchyma. The pathway and extent of hepatocellular dysfunction determine the specific manifestations of liver disease. The outcomes to be anticipated when normal hepatic functions fail are described later.
Some consequences of liver disease, particularly of cirrhosis, are best understood in terms of what we know about hepatic blood flow. Of greatest clinical importance are the existence under normal circumstances of a low-pressure portal venous capillary bed throughout the liver parenchyma and the functional zonation of portal blood flow.
When pathologic processes (eg, fibrosis) result in elevation of the normally low intrahepatic venous pressure, blood backs up and a substantial fraction of it finds alternative routes back to the systemic circulation, bypassing the liver. Thus, blood from the GI tract is, in effect, filtered less efficiently by the liver before entering the systemic circulation. The consequences of this portal-to-systemic shunting are loss of the protective and clearance functions of the liver, functional abnormalities in renal salt and water homeostasis, and a greatly increased risk of GI hemorrhage from the development of engorged blood vessels carrying venous blood bypassing the liver (eg, esophageal, gastric, umbilical varices, etc).
Even in the absence of any intrinsic parenchymal liver disease, portal-to-systemic shunting of blood can produce or contribute to encephalopathy (altered mental status resulting from failure to clear poisons absorbed from the GI tract), GI bleeding (resulting from esophageal varices), and malabsorption of fats and fat-soluble vitamins (caused by loss of enterohepatic recirculation of bile), with associated coagulopathy. In Table 14–4, the syndromes observed in liver disease are categorized as being a consequence of hepatocyte dysfunction, portal-to-systemic shunting, or both.
| Syndromes of Aberrant Function in Liver Disease | Hepatocellular Dysfunction | Portal-to-Systemic Shunting |
|---|---|---|
| Energy metabolism and substrate conversion | ||
| Alcoholic hypoglycemia | ✓ | |
| Alcoholic ketoacidosis | ✓ | |
| Hyperglycemia | ✓ | |
| Familial hypercholesterolemia | ✓ | |
| Hepatic encephalopathy | ✓ | ✓ |
| Fatty liver | ✓ | |
| Solubilization, transport, and storage function | ||
| Reactions to drugs | ✓ | |
| Drug sensitivity | ✓ | ✓ |
| Steatorrhea | ✓ | ✓ |
| Fat-soluble vitamin deficiency | ✓ | ✓ |
| Coagulopathy | ✓ | ✓ |
| Protein synthetic function | ||
| Edema due to hypoalbuminemia | ✓ | |
| Protective and clearance functions | ||
| Hypergammaglobulinemia | ✓ | |
| Hypogonadism and hyperestrogenism | ✓ | ✓ |
| Renal dysfunction | ||
| Sodium retention | ✓ | |
| Impaired water excretion | ✓ | |
| Impaired renal concentrating ability | ✓ | |
| Deranged potassium metabolism | ✓ | |
| Prerenal azotemia | ✓ | |
| Acute kidney injury | ✓ | |
| Glomerulopathies | ✓ | |
| Impaired renal acidification | ✓ | |
| Hepatorenal syndrome | ✓ | |
The fact that hepatocytes in the different zones of the acinus “see” blood in a particular sequence has great pathophysiologic significance. Because zone 1 hepatocytes see blood that has just left the portal venule or hepatic arteriole, they have access to the highest concentrations of various substances, both good (eg, oxygen and nutrients) and bad (eg, drugs and toxins absorbed from the GI tract). Zone 2 hepatocytes receive blood containing less of these substances, and zone 3 hepatocytes are bathed in blood largely depleted of them. However, zone 3 hepatocytes see the highest concentrations of products (eg, drug metabolites) released into the bloodstream by hepatocytes of zones 1 and 2. Thus, direct poisons have their most severe impact on zone 1 hepatocytes, whereas poisons that are generated as a result of hepatic metabolism cause more damage to those of zone 3. Similarly, because sinusoidal blood around zone 3 has the lowest oxygen concentration, hepatocytes of this zone are at greatest risk of injury under conditions of hypoxia.
Whether as a result of hepatocyte dysfunction or portal-to-systemic shunting, failure of normal hepatic function underlies the clinical manifestations of liver disease. An understanding of these mechanisms offers insight into the probable causes of illness in a patient with acute or chronic liver disease.
A first category of altered liver function involves the intermediary metabolism of carbohydrates, fats, and proteins.
Severe liver disease can result in either hypoglycemia or hyperglycemia. Hypoglycemia results largely from a decrease in functional hepatocyte mass, whereas hyperglycemia is a result of portal-to-systemic shunting, which decreases the efficiency of postprandial extraction of glucose from portal blood by hepatocytes, thus elevating systemic blood glucose concentration.
Disturbance of lipid metabolism in the liver can result in syndromes of fat accumulation within the liver early in the course of liver injury. Perhaps this is because the complex steps in assembly of lipoprotein particles for export of cholesterol and triglycerides from the liver are more sensitive to disruption than the pathways of lipid synthesis. Such disruption results in a buildup of fat that cannot be exported in the form of VLDL.
In certain chronic liver diseases such as primary biliary cirrhosis, bile flow decreases as a result of destruction of bile ducts. The decrease in bile flow results in decreased lipid clearance via bile, with consequent hyperlipidemia. These patients often develop subcutaneous accumulations of cholesterol termed xanthomas.
Any disturbance of protein metabolism in the liver can result in a syndrome of altered mental status and confusion known as hepatic encephalopathy. As with carbohydrate metabolism, altered protein metabolism can result from either hepatocyte failure or portal-to-systemic shunting, with the net effect of elevation of blood concentrations of centrally acting toxins, including ammonia generated by amino acid metabolism.
The clinical significance of bile synthesis can be seen in the prominence of cholestasis—failure to secrete bile—in many forms of liver disease. Cholestasis can occur as a result of extrahepatic obstruction (eg, from a gallstone in the common bile duct) or selective dysfunction of the bile synthetic and secretory machinery within the hepatocytes themselves (eg, from a reaction to certain drugs). The mechanisms responsible for cholestatic drug reactions are not well understood. Regardless of the mechanism, however, the clinical consequences of severe cholestasis may be profound: A failure to secrete bile results in a failure to solubilize substances such as dietary lipids and fat-soluble vitamins, resulting in malabsorption and deficiency states, respectively. Retained bile salts are also cytotoxic, but in the setting of cholestasis hepatocytes adapt to decrease uptake of bile salts by downregulating Na+-bile acid cotransporter while maintaining bile salt excretion. As a result, hepatic necrosis is minimized in predominantly cholestatic syndromes, with the typical laboratory findings of minimally elevated levels of AST and ALT in the presence of marked jaundice and high levels of bilirubin. However, prolonged exposure to bile salts in chronic cholestatic diseases such as primary biliary cirrhosis leads to portal tract cytotoxic injury and inflammation, leading eventually to fibrosis and cirrhosis.
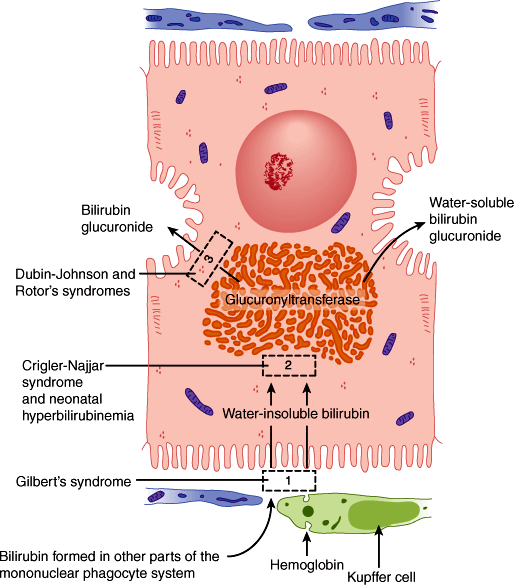
The solubilization function of bile works both to excrete and to absorb substances. Thus, in cholestasis, endogenous substances that are normally excreted via the biliary tract can accumulate to high levels. One such substance is bilirubin, a product of heme degradation (Figure 14–7). The buildup of bilirubin results in jaundice (icterus), which is a yellow discoloration of the sclera and skin. In the adult, the most significant feature of jaundice is that it serves as a readily monitored index of cholestasis, which may occur alone or with other abnormalities in hepatocyte function (ie, as part of the presentation of acute hepatitis). In the neonate, however, elevated bilirubin concentrations can be toxic to the developing nervous system, producing a syndrome termed kernicterus.
Figure 14–7
The secretion of bilirubin. This water-insoluble compound is derived from the metabolism of hemoglobin in macrophages of the mononuclear phagocyte system. Glucuronyl transferase activity in the hepatocytes causes bilirubin to be conjugated with glucuronide in the smooth endoplasmic reticulum, forming a water-soluble compound. Accumulation of bilirubin and bilirubin glucuronide in the tissues produces jaundice. Several defective processes in the hepatocytes can cause diseases that produce jaundice: a defect in the capacity of the cell to trap and absorb bilirubin (rectangle 1), the inability of the cell to conjugate bilirubin because of a deficiency in glucuronyl transferase (rectangle 2), or problems in the transfer and excretion of bilirubin glucuronide into the biliary canaliculi (rectangle 3). One of the most frequent causes of jaundice, however—unrelated to hepatocyte activity—is the obstruction of bile flow as a result of gallstones or tumors of the pancreas. This causes jaundice primarily as a result of accumulation of bilirubin glucuronide in the tissues. (Redrawn, with permission, from Junqueira LC et al, eds. Basic Histology, 10th ed. McGraw-Hill, 2003.)
Similarly, cholesterol is normally excreted either by conversion into bile acids or by forming complexes, termed micelles, with preexisting (recycled) bile acids. In cholestasis, the resultant buildup of bile acids can lead to their deposition in the skin. This is believed to cause intense itching, or pruritus. Data suggest that, in at least some patients, cholestasis results in altered levels of endogenous opioids. Instead of skin deposition of bile acids altered endogenous opioid-mediated neurotransmission may be responsible for pruritus. Disorders of bile production are a basis for the formation of cholesterol gallstones. Nevertheless, as mentioned, other hepatocyte functions are often relatively well preserved in the face of significant cholestasis. The syndromes that produce jaundice are summarized in Table 14–5.
| Blood Tests | ||||||
|---|---|---|---|---|---|---|
| Type of Jaundice | Hct | Unconjugated Bilirubin (Indirect) | Conjugated Bilirubin (Direct) | Alkaline Phosphatase | Aminotransferases | Cholesterol |
| Hemolytic | ↓ | ↑ | N | N | N | N |
| Hepatocellular | ||||||
| N | ↑ | N | N | N | N |
| N | |||||



