Chapter Five
Literature Evaluation II: Beyond the Basics
Learning Objectives
After completing this chapter, the reader will be able to
• Describe examples of other study designs besides the basic controlled clinical trial.
• Discuss the potential utility, limitations, and questions to ask when evaluating other study designs.
• Describe the characteristics of various observational trial designs.
• Differentiate between the three types of literature reviews: narrative (nonsystematic) review, systematic review, and meta-analysis.
• Describe common quality-of-life (QOL) measures used in health outcomes research and discuss the appropriate use of these measures in the medical literature.
• Discuss common issues encountered in dietary supplement (botanical and nonbotanical) medical literature.
• Describe how to efficiently and effectively evaluate the available evidence associated with a clinical question and categorize the quality of that evidence to develop a recommendation/clinical decision.
![]()
Key Concepts
![]() Although the randomized controlled trial is the most frequently used study design for clinical research, several other designs are used in specific situations, such as investigating rare outcome incidences, studying equivalency/noninferiority between drugs, or minimizing patient exposure to new drugs with inadequate efficacy.
Although the randomized controlled trial is the most frequently used study design for clinical research, several other designs are used in specific situations, such as investigating rare outcome incidences, studying equivalency/noninferiority between drugs, or minimizing patient exposure to new drugs with inadequate efficacy.
![]() Observational study designs offer an alternative to interventional trials and are used in specific situations, such as when large populations must be followed over extended periods of time. Results from these trials only allow associations to be formed rather than true cause-and-effect relationships.
Observational study designs offer an alternative to interventional trials and are used in specific situations, such as when large populations must be followed over extended periods of time. Results from these trials only allow associations to be formed rather than true cause-and-effect relationships.
![]() Case studies, case reports, and case series are reports describing patient or patient group exposure to a drug or technology and can be valuable to record preliminary findings that lead to further study. A key characteristic to these reports is the lack of a control or comparison group.
Case studies, case reports, and case series are reports describing patient or patient group exposure to a drug or technology and can be valuable to record preliminary findings that lead to further study. A key characteristic to these reports is the lack of a control or comparison group.
![]() Survey research is information gathered from an identified group with conclusions drawn and applied to a larger population. This gathered information is considered either descriptive (such as opinions and attitudes) or explanatory (such as explaining a cause and effect) in nature and the validity of the results depends on quality of the study’s internal rigor.
Survey research is information gathered from an identified group with conclusions drawn and applied to a larger population. This gathered information is considered either descriptive (such as opinions and attitudes) or explanatory (such as explaining a cause and effect) in nature and the validity of the results depends on quality of the study’s internal rigor.
![]() Meta-analyses are the only type of review providing new quantitative data derived from combining the results of each study in the meta-analysis and performing a statistical analysis on that data set. The overall reliability of a meta-analysis is ultimately dependent on the quality of the individual studies, homogeneity between these studies, and the appropriateness of the analysis.
Meta-analyses are the only type of review providing new quantitative data derived from combining the results of each study in the meta-analysis and performing a statistical analysis on that data set. The overall reliability of a meta-analysis is ultimately dependent on the quality of the individual studies, homogeneity between these studies, and the appropriateness of the analysis.
![]() The value assigned to quality and quantity of life affected by many different variables including disease, injury, treatment, or policy is termed health-related quality of life (HR-QOL) and is used to assist in decision making regarding interventions such as procedures and pharmacotherapy.
The value assigned to quality and quantity of life affected by many different variables including disease, injury, treatment, or policy is termed health-related quality of life (HR-QOL) and is used to assist in decision making regarding interventions such as procedures and pharmacotherapy.
![]() The principles and criteria used to analyze the quality of drug literature are used to analyze dietary supplement (DS) literature; however, unique additional points such as standardization and purity must be considered.
The principles and criteria used to analyze the quality of drug literature are used to analyze dietary supplement (DS) literature; however, unique additional points such as standardization and purity must be considered.
![]() An understanding of strengths and limitations inherent with each study design is essential to determine the overall quality of the evidence produced. Those trial designs with a high level of quality provide the most reliable evidence and that translates into the strongest recommendation/clinical decision.
An understanding of strengths and limitations inherent with each study design is essential to determine the overall quality of the evidence produced. Those trial designs with a high level of quality provide the most reliable evidence and that translates into the strongest recommendation/clinical decision.
Introduction
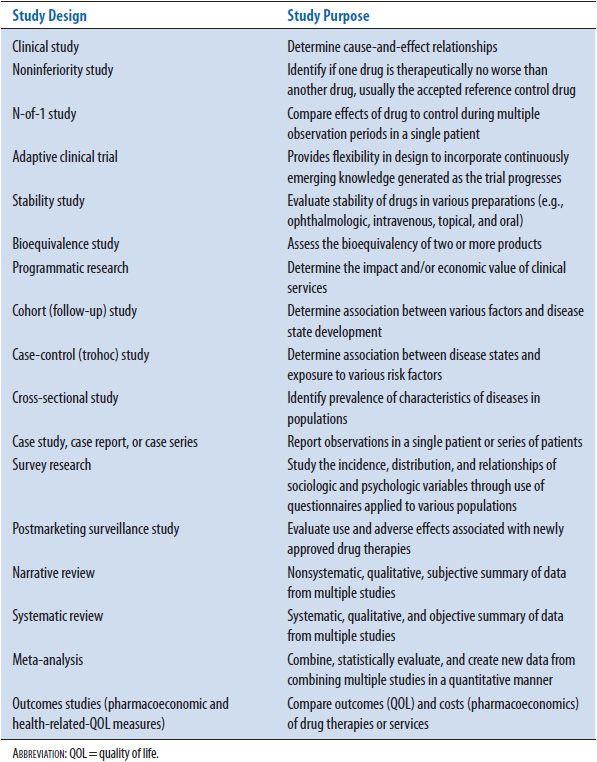
![]() Although the randomized controlled trial is the most frequently used study design for clinical research, several other designs are used in specific situations, such as investigating rare outcome incidences, studying equivalency/noninferiority between drugs, or minimizing patient exposure to new drugs with inadequate efficacy. Principles that apply to randomized controlled trials (see Chapter 4) also apply to other types of study designs. There are situations where other research designs are more effective in answering specific questions or providing the only data available to answer the questions. For example, there has been a tremendous increase in the use of noninferiority study design to establish a drug’s position in therapy.1 Typically, a superiority trial design is used in this instance. The issue arises when no statistically significant difference in efficacy between the new drug and control or reference drug (often considered to be standard treatment) is shown. The only conclusion to be made at that time is that the new drug is not superior to the reference drug. When a noninferiority trial design is used and the results confirm that the new drug is noninferior to the reference drug, a conclusion can be made that the new drug is “no worse” than the reference drug regarding efficacy. If noninferiority is established, the same data can be analyzed for superiority of the new drug over the reference drug. If no statistically significant difference in efficacy is shown from this analysis and, therefore, no superiority is established, the researchers at least know the new drug is still no worse than the reference drug. As another example, adaptive clinical trial design has become more common in assisting to establish efficacy for a particular drug. Adapting the number of patients, eligibility criteria, drug dose, or randomization allocation can significantly improve the efficiency of drug development and provide a more realistic clinical environment to test a new drug. Table 5–1 lists commonly encountered biomedical literature.
Although the randomized controlled trial is the most frequently used study design for clinical research, several other designs are used in specific situations, such as investigating rare outcome incidences, studying equivalency/noninferiority between drugs, or minimizing patient exposure to new drugs with inadequate efficacy. Principles that apply to randomized controlled trials (see Chapter 4) also apply to other types of study designs. There are situations where other research designs are more effective in answering specific questions or providing the only data available to answer the questions. For example, there has been a tremendous increase in the use of noninferiority study design to establish a drug’s position in therapy.1 Typically, a superiority trial design is used in this instance. The issue arises when no statistically significant difference in efficacy between the new drug and control or reference drug (often considered to be standard treatment) is shown. The only conclusion to be made at that time is that the new drug is not superior to the reference drug. When a noninferiority trial design is used and the results confirm that the new drug is noninferior to the reference drug, a conclusion can be made that the new drug is “no worse” than the reference drug regarding efficacy. If noninferiority is established, the same data can be analyzed for superiority of the new drug over the reference drug. If no statistically significant difference in efficacy is shown from this analysis and, therefore, no superiority is established, the researchers at least know the new drug is still no worse than the reference drug. As another example, adaptive clinical trial design has become more common in assisting to establish efficacy for a particular drug. Adapting the number of patients, eligibility criteria, drug dose, or randomization allocation can significantly improve the efficiency of drug development and provide a more realistic clinical environment to test a new drug. Table 5–1 lists commonly encountered biomedical literature.
TABLE 5–1. COMMONLY ENCOUNTERED BIOMEDICAL LITERATURE
To effectively determine the quality of these trials, the practitioner must first evaluate major considerations that apply to the design. One approach uses a list of essential study components referred to as the Ten Major Considerations Checklist.2 The Ten Major Considerations include
1. Power of the study set/met (to estimate adequate sample size that will identify a difference between groups if one truly exists)?
2. Dosage/treatment regimen appropriate?
3. Length of study appropriate to show effect?
4. Inclusion criteria adequate to identify target study population?
5. Exclusion criteria adequate to exclude patients who may be harmed in study?
6. Blinding present?
7. Randomization resulted in similar groups?
8. Biostatistical tests appropriate for type of data analyzed?
9. Measurement(s) standard/validated/accepted?
10. Author’s conclusions are supported by the results?
Once this is completed, additional considerations unique to a specific study design discussed in this chapter must be evaluated. For example, it is necessary to determine the similarity of studies used to conduct a meta-analysis since the results of each study will be combined and new data created and analyzed.
The purpose of this chapter is to familiarize the health care practitioner with these unique study designs. In addition, specific considerations unique to each design and in addition to the Ten Major Considerations are identified for the practitioner to evaluate (see Appendix 5–1). Finally, a process is provided to bring all the results of trials focused on a particular clinical question together and develop a recommendation/clinical decision based on this evidence. For these reasons, this chapter can be extremely valuable and useful to anyone who depends on the medical literature to provide answers to their daily clinical challenges.
This chapter covers the variations on randomized clinical trial design (noninferiority, N-of-1, and adaptive clinical trials), observational trial design (cohort, case-control, and cross-sectional), and uncontrolled study designs (case studies, case series, and case reports). Specific types of studies such as bioequivalency, postmarketing surveillance, and programmatic research are discussed. Differences between narrative (qualitative), systematic (qualitative), and meta-analysis (quantitative) reviews are addressed. A discussion of the specialized area of health outcomes research is included to introduce concepts directly related to patient quality-of-life (QOL) trials. A section on evaluation of dietary supplement (DS) medical literature including issues specific to these trials and common methodological flaws is provided.
Good literature evaluation skills and application of a systematic evidence-based medicine (EBM) process provide the foundation that allows clinicians to make the best recommendations and decisions.3 In light of rapidly emerging evidence and busy practitioner schedules, an understanding of inherent strengths and limitations associated with various types of clinical trial design provides a powerful clinical tool. In general, those trials with a greater number of identified strengths than limitations, especially if those limitations have a minimal effect on the study results, provides a high level of reliability. Using these high-quality trials that represent the best evidence, a stronger recommendation/clinical decision can be made. This in turn ensures the highest level of patient care, whether for one specific patient or large patient populations.
Beyond the Basic Controlled Trial
NONINFERIORITY TRIALS
Description
Randomized, controlled clinical trials are utilized to determine one of three different outcomes between comparative drugs: superiority, equivalency, or noninferiority (NI).4 All three trial designs involve a new test drug compared to the accepted active control or standard of care drug. With superiority trials, the aim is to determine that one drug is superior to the other.5 In contrast, equivalency trials are designed to determine if the new drug is therapeutically similar to the control drug. This trial design is used primarily to determine bioequivalence between two drugs. Noninferiority trials seek to show that any difference between two treatments is small enough to conclude the test drug has “an effect that is not too much smaller than the active control”6 also referred to as the reference drug or standard treatment.1,6–9 This section will focus on description, interpretation, and evaluation of NI trials.
There has been a dramatic increase in rate of the NI studies since 1999.1 Along with this increase has come an understanding of several potential study design issues including choice of reference drug, at what point the test drug’s treatment effect will be considered inferior to the reference drug, and optimal analytical approaches, therefore, requiring rigor in design and conduct of these trials.10,11 An NI study design is often considered when superiority of a test drug over a reference drug is not anticipated.12 In this case, the objective is showing the test drug to be statistically and clinically not inferior to the reference drug based on efficacy, yet the test drug could offer other potential advantages related to safety, tolerability, convenience, or cost.7,8 These ancillary benefits can justify use of the test drug to replace standard treatment. NI trials are also considered when the use of a superiority trial would be considered unethical.6 For example, it is unethical to use a placebo when there is available effective treatment that possesses an important benefit, such as preventing death or irreversible injury to the patient. For this reason, NI trials may provide an alternative method to using superiority trials for meeting United States (U.S.) Food and Drug Administration (FDA) marketing approval requirements.6 Between 2002 and 2009, 14% of the New Drug Applications (NDAs) approved contain evidence from pivotal NI trials.13 Rather than going through the process of showing a new drug is therapeutically superior to the reference drug treatment or a placebo, pharmaceutical companies are choosing to use an NI design. This is a developmental strategy to confirm the new drug has a valid therapeutic effect and that the drug’s effect is not, at minimum, worse than the reference drug. Often the anticipated key differentiating factor for the test drug in this situation is improved safety profile. For example, a new drug for cardiac arrhythmias is anticipated to have similar efficacy but less serious side effects than the standard treatment. It should be noted that NI design is not recommended when the reference drug treatment effect is not consistently superior to placebo (both statistically and clinically).14
Establishing the NI Margin
The NI margin is an important component of an NI trial and is a prespecified amount used to show the test drug’s treatment effect is not worse than the reference drug by more than this specific degree.15 A combination of statistical reasoning and clinical judgment is required to best determine the NI margin.7,8,16,17 Another way to think of the NI margin is the extent to which the test drug’s effect can be worse than the reference drug, but still be considered no worse.18 For instance, if the reference drug’s minimal treatment effect is determined to be a 5 mmHg drop in diastolic blood pressure compared to placebo, then the test drug could not be worse than this 5 mmHg change to be considered noninferior to the reference drug. In other words, if the results from the NI trial showed the test drug’s treatment effect to be 4 mmHg, then the test drug would be considered potentially inferior to the reference drug. The FDA draft guideline document defines the NI margin as the largest clinically acceptable difference (degree of inferiority) of the test drug compared to the reference drug.6 Methods to determine the NI margin have been proposed; however, concern exists with the subjectivity and potential bias associated with any method that relies on an indirect comparison of historical data rather than a true placebo arm built into the NI study.19–22
A simple method to determine the NI margin is to look at the confidence interval (CI) for the reference drug treatment effect (reference drug – placebo = treatment effect). This treatment effect is best determined from examining the historical reference drug versus placebo-controlled trials. A meta-analysis of these historical placebo-controlled trials is preferred to obtain the most accurate overall treatment effect when several studies are involved.8 The CI around the mean reference drug treatment effect is used to establish the NI margin (see Chapter 8 for an understanding of CIs). The lower bound of that CI represents the smallest expected reference drug treatment effect. For example, if the CI around the mean reference drug treatment effect is 3 to 14 mmHg diastolic blood pressure, then the lower bound of that CI would be 3 mmHg. This lower bound can be used to set the NI margin; however, a 5 mmHg change in diastolic blood pressure could be considered the smallest clinically significant change. Given this information, the NI margin can be set at 5 mmHg.
Interpretation of Results
The NI design focuses on the mean treatment difference between the test drug and the reference drug (see Figure 5–1). To determine NI, the CI around the mean treatment difference between the test drug and the reference drug is compared against the NI margin. If the CI around the mean treatment difference includes (or crosses) the NI margin, then the possibility of the test drug being inferior to the reference drug exists (inconclusive), and thus, the test drug fails to exhibit noninferiority (see Scenario A in Figure 5–1). The test drug would be determined inferior if the entire CI around the mean treatment difference was located on the inferior side of the NI margin (see Scenario B in Figure 5–1). However, if the CI around the mean treatment difference does not include or cross the NI margin, then a conclusion can be made that the test drug is noninferior to the reference drug (see Scenarios C and D in Figure 5–1).
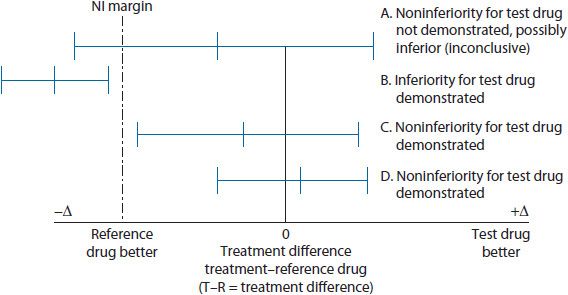
Figure 5–1. Noninferiority trial design concept.
For example, an NI study of a new antihypertensive agent (test drug) compared to reference drug is published in the literature. The overall treatment effect of the reference drug compared to previous placebo-controlled studies is a reduction of 12 mmHg (CI 6 to 15 mmHg) for a sitting diastolic blood pressure. The lower bound of that CI = 6 mmHg. This lower bound or some percentage of that taking into consideration clinical significance can be used to help set the NI margin. In this case, the lower bound of the CI of 6 mmHg treatment effect is easily considered a clinically significant difference or change in sitting diastolic blood pressure. Given this, the NI margin is established as 6 mmHg sitting diastolic blood pressure. In other words, when the NI trial is completed the test drug’s lower bound of the CI associated with the mean treatment difference cannot include or cross the set NI margin of 6 mmHg on the graph (see Figure 5–2). If the test drug’s lower bound of the CI includes (or crosses) the NI margin, this would suggest the test drug failed to exhibit noninferiority (possibly inferiority) to the reference drug (see Scenario A in Figure 5–2). This situation would be considered an inconclusive result unless the entire CI for the mean treatment difference is located on the inferiority side of the NI margin of 6 mmHg, confirming inferiority. Upon completion of the example antihypertensive NI study, the actual results confirm noninferiority of the new antihypertensive agent based on the lower bound of the CI interval for the test drug treatment effect not including (or crossing) the NI margin (see Scenario B in Figure 5–2). In the case where noninferiority is determined, three assumptions are made: (1) test drug exhibits noninferiority to the reference drug, (2) test drug performs better than placebo if placebo was included as a comparator group, and (3) test drug may offer ancillary benefits and these could include safety, tolerability, convenience, or cost.7
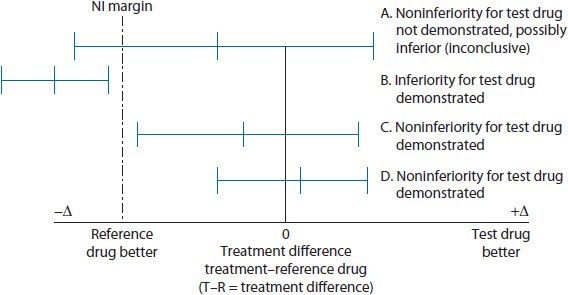
Figure 5–2. Noninferiority trial design example.
If an NI margin and/or CI are not provided, then noninferiority can still be determined if an alpha and p-value have been given. To use p-values for this purpose there must be an understanding that statistical rationale for NI trials differs from that used with superiority trials.23,24 Specifically, defining parameters for the null and alternative hypotheses (see Chapters 4 and 8) are different from those used with superiority trials. In fact, the definitions for the two hypotheses are essentially switched or reversed and relabeled compared to superiority trial hypotheses7,8 (see Figure 5–3). The null hypothesis for a superiority trial postulates there is no statistically significant difference between treatment groups, and the alternative hypothesis states there is a statistically significant difference between the treatment groups. With NI trials, the null hypothesis states the test drug fails to exhibit noninferiority to the reference drug or at the very least the results are inconclusive for noninferiority versus inferiority.12 The alternative hypothesis states that the test drug is noninferior to the reference drug (not significantly worse). With NI trials, the null hypothesis must be rejected in favor of the alternative hypothesis to conclude noninferiority for the test drug. This requires a p-value less than the set alpha value. For example, an alpha of 0.05 indicating the significance level, a p-value <0.05 would indicate NI for the test drug. If the null hypothesis is not rejected, then the test drug fails to exhibit noninferiority. In this case the p-value would be equal to or greater than the set alpha value. For example, a p-value is provided rather than an NI margin and/or CI around the mean treatment difference.

Figure 5–3. Differences in defining parameters for null and alternative hypotheses.
Some controversy exists whether an NI trial alone can show superiority of the test drug over the reference drug.5,23,25 Once NI is established, superiority can be tested using the results for the primary outcome measure(s).7,8 According to guidance from the FDA, an NI study can be designed to first test for noninferiority with a predetermined NI margin, and if successful proving noninferiority, the data can be analyzed for potential superiority of the test drug. Note the importance of the sequential process of first proving noninferiority, then analyzing for superiority. For instance, if the new antihypertensive NI trial discussed earlier provided results that showed the test drug to be noninferior, then an analysis of superiority would be appropriate and acceptable. This should be stated in the original noninferiority trial protocol. Superiority can be tested prior to NI, but the NI margin must be stated a priori to avoid potential manipulation of the margin. Adjusting the NI margin to avoid the CI from crossing over would give a false NI result and is unacceptable. In addition, seeking the conclusion of NI from a failed superiority trial (one that shows no statistically significant difference between treatment groups) is not acceptable.6,26 The only conclusion that can be made from this scenario is that the test drug is not superior to the reference drug. If a noninferiority conclusion is anticipated, an NI study design should be the initial choice.
Evaluation
Several study design characteristics must be considered when evaluating the quality of an NI trial.5 The Ten Major Considerations Checklist discussed in the Introduction section is used to identify specific major strengths and limitations.2
Of significant importance is how the NI margin used to determine noninferiority is defined.23,27 Correctly determining this margin is considered the greatest challenge in the design and interpretation of NI trials.6 If noninferiority is established for the test drug, further concluding the test drug is effective can only be made if the reference drug’s efficacy has been confirmed with high-quality clinical trials against placebo. This is referred to as assay sensitivity.6,21,26 The NI margin is set using historical studies to determine the actual effect of the reference drug. Two approaches to determine the NI margin are most frequently referenced: the fixed margin approach and the synthesis approach. Both these approaches use historical data. The fixed-margin approach is preferred by the FDA since that approach is more conservative than the synthesis approach.28
Historical evidence of sensitivity to drug effects (HESDE) is a term used to describe appropriately designed and conducted past trials using the reference drug and regularly exhibiting the reference drug to be superior to placebo. Meta-analytic methods can be used to develop more precise estimates of reference drug effect when several studies are available. When multiple studies exist, all studies should be considered to avoid overestimating the reference drug’s effect.6 Only after a determination is made that these past studies are similar in design and conduct compared to the NI trial regarding features that could alter the effect size can the HESDE be used to select the NI margin. In other words, the historical studies and new NI study should be as identical as possible regarding important characteristics. This is called the constancy assumption.12,21 Note that establishing efficacy for the test drug using an NI trial design is seriously complicated by assay sensitivity and constancy assumptions that cannot be completely verified, leading to potentially inflated Type I error rate (see Chapters 4 and 8).28 The concern is that approval of an ineffective therapy can result with inflated Type I error rates since the test drug is falsely determined to be noninferior to the reference drug.
Another concern called biocreep or placebo creep can exist that complicates using an active control to confirm efficacy as is done with an NI trial.12,21 Biocreep is the phenomenon where a somewhat inferior test drug is chosen as the reference drug for a future generation of NI trials. When this occurs with generation after generation of NI trials, the future reference drug becomes no better than placebo.
As mentioned earlier, but important enough to reiterate, setting the NI margin post hoc can be interpreted as potential manipulation by investigators to show desired results. For this reason, it is critical that the NI margin be set prospectively before the beginning of the trial. This is considered acceptable practice. For all of these reasons, the investigators should provide a detailed description of how the NI margin was determined.
The type of analysis used should also be determined prospectively. For the purposes of NI study analysis, a per-protocol (PP) analysis where only the patients that completed the study are included is preferred over an intention-to-treat (ITT) analysis (see Chapters 4 and 8).5,23 The ITT analysis includes all patients that were randomized to treatment regardless of whether they completed the study duration. With the ITT analysis, smaller observed treatment effects can result since patients may not have stayed in the study long enough to see the maximum effect of the test drug. Using an ITT analysis with an NI trial can significantly increase the risk of falsely claiming noninferiority due to the potential of smaller observed treatment effects.8,26 For this reason, when an ITT analysis is used, a PP analysis is also used to cross-validate the ITT analysis.8,29 Note that the PP analysis can be invalidated based on significant variation in dropout rates between treatment groups.29 For example, a noninferiority study is completed but there is an excessively large dropout rate due to safety and lack of return to the clinic visits. This provides a much smaller PP population to be analyzed. If that PP population is less than the required number of patients to meet the set power, then questions arise if no statistical significance (p < 0.05 if alpha set at 0.05) is shown. This lack of statistical significance shown could be real and the test drug would fail to exhibit noninferiority (fail to reject the null hypothesis). Since power was not met the result could represent a Type II error (no statistical significance is shown when there really is one and, therefore, the test drug is noninferior to the reference drug) that invalidates the PP analysis (see Chapters 4 and 8 for explanation of Type II errors). The FDA and others suggest conducting and reporting both a PP and ITT analyses.6,9,29,30 Any differences in results between these two separate analyses should be examined closely by the reader.
Case Study 5–1: Noninferiority Trial Design
A 14-day multicenter randomized double-blind double-dummy noninferiority (NI) trial was conducted to compare short-term efficacy of a generic alginate/antacid to omeprazole on GERD symptoms in general practice. Alginate is the salt of alginic acid and the combination of the alginic acid and bicarbonate creates a barrier which prevents stomach acid from refluxing back up into the esophagus. In addition, safety was compared between the two drugs throughout the study. The primary outcome measure was the mean time to onset of the first 24-hour heartburn-free period after initial dosing. Based on the most recent study, an NI margin was set at 0.5 days. An NI margin of 0.5 days means noninferiority of the alginate/antacid would be shown by a clinically relevant value of 0.5 days less than for omeprazole. In other words, the mean difference in onset of the first 24-hour heartburn-free period between the alginate/antacid and omeprazole with the corresponding 95% CI was used for the noninferiority test. If the lower limit of the CI was within 0.5 days and did not cross this NI margin, then the noninferiority hypothesis was confirmed for the alginate/antacid product. Intent-to-treat and per protocol analyses were used. Safety was assessed by the number of reported adverse drug reactions for each drug.
• Identify the null hypothesis and alternative hypothesis for this noninferiority study.
• When setting the NI margin, what specific things did the investigators need to take into consideration?
• Did the investigators use the appropriate analysis type for an NI study?
• What is your conclusion if the 95% CI around the mean treatment difference of onset of first 24-hour heartburn-free period between alginate/antacid and omeprazole does not include or cross over the established NI margin?
• If the 95% CI around the mean treatment difference of onset of first 24-hour heartburn-free period between alginate/antacid and omeprazole includes or crosses over the established NI margin, what is your conclusion?
• After showing noninferiority for the two drugs, the investigators performed a superiority analysis on the data. What is your reaction to this second analysis?
![]()
NI trials typically have a larger sample size compared to superiority trials since the NI margin is frequently smaller than the treatment difference anticipated with superiority trials.26,31 Sample size determination before the study is started can be difficult since variance of the estimated treatment effect and event rate can be unknown for the test drug at this point. When this issue is present for superiority studies, an independent data monitoring group/committee is formed to examine the blinded information during the trial (interim analysis). This committee is looking for the occurrence of an unexpectedly low event rate so an upward adjustment in sample size can be proposed. This same approach to sample size reassessment and adjustment is applied to NI trials.32,33 A clear explanation of how the blinded information was handled and how the independent data monitoring committee (IDMC) was conducted should be provided in the article for evaluation.
Reporting NI trials in the literature is still not consistent and this can often be the biggest issue to evaluating and interpreting trial results for quality.34 The Consolidated Standards of Reporting Trials (CONSORT) statement, first published in 1996 and then updated in 2010 to improve randomized controlled trial reporting in the medical literature.35,36 Extensions to the CONSORT statement that specifically address reporting of NI trials have been proposed.5,8,37 Specifically, these points include:
• Rationale for conducting trial
• Research objective(s)
• Clearly defined primary and secondary outcome measures
• Statistical methods used to evaluated objective(s)
• Methods used for sample size determination
• Inclusion/exclusion criteria comparable to previous trials using the active comparator (reference drug/devise/procedure)
• Description accounting for patients that are screened, randomly allocated, violated protocol, and completing study
• Explanation of how NI margin was determined
• Rationale for choosing comparator with historical study results confirming efficacy of this comparator
• Explanation of interim analyses and study discontinuation rules
• Interpretation of the real and estimated treatment exposures
• Explanation of the impact withdrawals or crossovers could have had on length of exposure to treatment assigned
• Summary of results for each primary and secondary outcome measure providing estimated effect size and precision (e.g., 95% CI)
• Interpretation of results considering the noninferiority hypothesis, potential biases, concerns with any multiple analyses.
As mentioned previously, an attempt to rescue a failed superiority study that concludes the results really demonstrate NI should not be accepted.23 Unfortunately, this situation is more common than would be expected.38 The plan to determine NI of a test drug compared to the reference drug must be established prospectively utilizing the proper study design to test for NI. In other words, lack of superiority in a superiority trial design does not equal NI.
Case Study 5–2: Noninferiority Trial Design
A 2-year double-blind randomized parallel group noninferiority (NI) trial was conducted to compare the safety and efficacy of a dipeptidyl peptidase-4 inhibitor (linagliptin) to a commonly used sulfonylurea (glimepiride) in Type 2 diabetic patients inadequately controlled on metformin. Patients already on metformin, but not adequately controlled, were randomized to receive either linagliptin or glimepiride for 104 weeks. Power was set and met with a sample size of over 707 patients per treatment group. The primary efficacy endpoint was change in hemoglobin A1c (HbA1c) from baseline to week 104. The mean difference between treatment groups in change of HbA1c from baseline to week 104 was the primary outcome for noninferiority. Even though this was an NI study, no mention of an NI margin was made throughout the study. Hypotheses based on noninferiority were established and a p-value of 0.03 was presented based on an alpha of 0.05. Safety was assessed by the number of reported adverse drug reactions for each drug.
• Identify the null hypothesis and alternative hypotheses for this noninferiority study.
• Since there was no NI margin, how do you determine if noninferiority exists between these two drugs? Did the investigators use the appropriate analysis type for an NI study?
• Let us say the p-value for the noninferiority evaluation is p = 0.063 with alpha set at 0.05. What is your conclusion? After showing noninferiority for the two drugs, the investigators performed a superiority analysis on the data. What is your reaction to this second analysis?
![]()
Appendix 5–1 contains a list of additional questions specific to NI studies. These specific questions are in addition to the standard questions used to evaluate other randomized clinical trials noted in Chapter 4 and using basic tools like the Ten Major Considerations Checklist discussed in the Introduction section.
N-OF-1 TRIALS
Description
The N-of-1 trial attempts to apply the principles of clinical trials, such as randomization and blinding, to individual patients.39 These trials are useful when the beneficial effects of a particular treatment in an individual patient are in doubt. It is advantageous if the treatment has a short half-life (allowing multiple crossover and washout periods without carryover effects—see Chapter 8 for further explanation) and is being used for symptomatic relief of a chronic condition.40 An N-of-1 trial can be used to determine whether a drug is effective in an individual patient. Taken as a whole, a group of N-of-1 trials can help identify characteristics that differentiate responders from nonresponders. Trials of multiple doses can identify the most effective dose and the clinical endpoints most influenced by the drug.41
An N-of-1 trial is similar to a crossover study conducted in a single patient who receives treatments in pairs (one period of the experimental therapy and one period of either alternative treatment or placebo) in random order.41 As described below, the study usually consists of several treatment periods that are continued until effectiveness is proven or refuted. Randomization to active drug or placebo, and blinding of the physician and patient to the treatment being administered, helps reduce treatment order effects (the order in which patients receive the treatments in the trial affects the results), placebo effects(therapeutic activity provided by administering a placebo), and observer bias (person collecting the data from the trial knows or has some idea what study drug each patient is receiving and, therefore, this may have some effect on the results reported). Desired outcomes are identified prior to initiation of the study to ensure that objective criteria that are meaningful to both the prescriber and the patient are used to assess treatment efficacy.40
N-of-1 trials may improve appropriate prescribing of drugs in individual patients. For example, carbamazepine may be an option for relief of pain in a patient with diabetic neuropathy, but definitive information on the efficacy of such treatment is limited. Therefore, investigators may conduct an N-of-1 trial to determine whether such therapy is useful in a particular patient. N-of-1 trials are especially useful when long-term treatment with a specific drug may result in toxicity and the prescriber wishes to determine whether benefits outweigh potential risks.40
The effectiveness of N-of-1 trials has been evaluated in a study.41,42 In this instance, of 57 N-of-1 trials completed, 50 (88%) provided a definite clinical or statistical answer to a clinical question, leading to the conclusion by the authors that N-of-1 trials were useful and feasible in clinical practice.42 Simply stated, the goal of an N-of-1 trial is to clarify a therapeutic management decision.42 Of 34 completed N-of-1 trials evaluated over a 2-year period, 17 (50%) were judged to provide definitive results (10 showed treatment to be effective, five showed treatment no better than no treatment, and two demonstrated harmful effects to the patients).43 The remaining 17 N-of-1 trials showed trends toward equivalence to control or actually favored placebo. Overall, clinician confidence in the therapy was found to increase or decrease depending on the direction of trial results.43
Evaluation
General evaluation requirements have been recommended for N-of-1 trials.43 Readers should determine whether the treatment target (or measure of effectiveness) was evaluated during each treatment period.44 This target should be a symptom or diagnostic test result, but must be directly relevant to the patient’s well-being (e.g., the visual analog scale for pain in the example of carbamazepine). Two other critical characteristics of an N-of-1 trial are that the symptom under investigation shows a rapid improvement when effective treatment is begun and that this improvement regresses quickly (but not permanently) when effective treatment is discontinued.44 The longer it takes to see a therapeutic effect provided by a drug, the longer the testing period of the trial. This also holds true for how long it takes for the therapeutic effect to disappear after stopping the drug; a longer washout period results in an overall increase in trial duration. The length of the treatment period is important to know. For those diseases that remain constant, this treatment period is easy to establish. When dealing with a disease that is not constant, such as multiple sclerosis, the treatment period must be long enough to observe an exacerbation of the condition. A general rule is that if an event occurs an average of once every X days, then a clinician needs to observe three times X days to be 95% confident of observing at least one event. One should ask if a clinically relevant treatment target is used and can it be accurately measured? It is advisable to measure symptoms or the patient’s QOL directly, with patients rating each symptom at least twice during each study period.
It is important to determine if sensible criteria for stopping the trial is established. Specification of the number of treatment pairs in advance strengthens the statistical analysis of the results and it has been advised that at least two pairs of treatment periods are conducted before the study is unblinded.41
N-of-1 trials provide more objective information than case reports or case studies because there is a control group, and are useful for providing definitive information for drug prescribing in individual patients. See Table 5–2 for a comparison of N-of-1 trials and case studies.
TABLE 5–2. COMPARISON OF N-OF-1 TRIALS AND CASE STUDIES
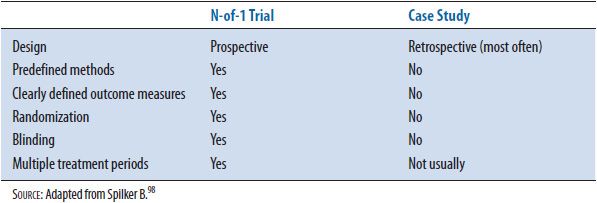
Appendix 5–1 contains a list of additional questions specific to N-of-1 studies. These specific questions are in addition to the standard questions used to evaluate other randomized clinical trials noted in Chapter 4 and using basic tools like the Ten Major Considerations Checklist discussed in the Introduction section.
ADAPTIVE CLINICAL TRIALS
Adaptive clinical trial (ACT) design has become a topic of great interest recently by both the pharmaceutical industry and the FDA.44 In simplest form, the ACT is known as a staged protocol or group sequential trial.45 One example of this group sequential design is called a 3+3 trial used in a Phase I trial to identify the maximum tolerated dose (MTD). Three patients start at a specific dose and if no toxicity is noted, a second group of three patients are given a higher dose. If one patient experiences a limiting toxicity, then a third group of three patients are given that same dose. From this third group, two or maybe all three patients experience toxicity, which leads to claiming that the previous lower dose is the MTD. This is one basic framework of the design that can be used for a specific purpose, dose response.
Key to this study design is that ACT provides the ability to use more adaptive sampling strategies. These strategies include response-adaptive designs for clinical trials where the emerging data or observations from the trial are used to make adjustments in the ongoing study.46 As illustrated in this example where an ACT design is incorporated into a dose-response study, emerging patient outcomes are utilized to adjust allocation of future enrolled patients or some other study design component.
Classical clinical trial design is rigidly structured to investigate a set number of variables and prevent additional variables from being introduced that will confound the results. For instance, a traditional, randomized, controlled trial stipulates inclusion and exclusion criteria to allow only a specific population of patients entering into the trial. The benefit of these strict criteria is a well-defined population to be studied that excludes patients predicted to be potentially harmed if entered into the trial. However, the disadvantage of such rigidity is the inability to make adjustments to the inclusion and exclusion criteria as the study progresses and new information comes from those patients who have completed the trial. If the investigators discover there is a specific patient group that responds to the treatment during the trial, adjustments to include only that patient population are not allowed. For that reason, a significant number of patients who are anticipated to not respond are exposed to a treatment that may cause severe side effects. ACT design provides enough flexibility to incorporate continuously emerging knowledge generated as a trial is carried out. In essence, the ACT design provides the researcher with an opportunity to change some study methodology when they identify things that may need to be done differently, such as using a different dose, different patients, or measuring different outcomes over a different period of time. Ideas from industry, academia, and regulatory agencies, such as the FDA Critical Path Initiative (http://www.fda.gov/ScienceResearch/SpecialTopics/CriticalPathInitiative/default.htm), are leading a movement toward ACT design as a potential alternative method to gather data for use in the FDA drug approval process.
The primary benefit of ACT design is that fewer patients are allocated to a less effective therapy, a greater number of responding patients can be monitored for safety of the effective therapy, and fewer patients overall may be required to determine statistical and clinical significance of the drug.47 Other potential benefits include a more efficient developmental pathway for the drug, patients benefit from effective therapies earlier, and prescribers have more information on patients most likely to benefit from the drug.
The majority of studies utilizing the ACT design have been dose-response trials used to identify the range of effective doses to be used in future efficacy confirmatory studies.47 A much smaller number of pivotal, efficacy confirming studies have used this design due to company concerns about FDA’s skepticism of ACT design including potential bias and increase in false-positive rates. With time, the ACT design is becoming more common in agreements made between the FDA and sponsors (also known as Special Protocol Assessments) for later stages of clinical development.
Higher level, more powerful, and complex ACTs require the use of Bayesian statistics that allow for much more flexibility than traditional statistical approaches can provide.46 Traditional statistical approaches determine the likelihood that the efficacy of a specific drug could have happened by chance. On the other hand, the Bayesian approach provides a probability (or relative likelihood) that the drug is effective. This approach estimates the relative likelihood by using new data as it is created from the ongoing study. The Bayesian approach continuously updates the relative likelihood of the subject being investigated.
Pfizer’s ASTIN trial (Acute Stroke Therapy by Inhibition of Neutrophils) is an example of how ACT methodology can be incorporated into a randomized, double-blind, placebo-controlled dose-response study.48 This trial represents the first time that computer-assisted, real-time learning was successfully implemented in a large international study to look at dose response effects. In this study, ACT design methodology was used to help determine the maximum effective dose and ensure the study drug’s best chance of showing efficacy. At the same time, the use of the ACT design decreased the exposure of patients to a drug that was not found to be efficacious in this particular disease state. The ACT study design is computationally and logistically complex, but today’s rapid data collection methods and computing power is adequate to meet the requirements. Software programs developed in-house by pharmaceutical industry statisticians are used to handle these complexities.
Studies based on the ACT design are evaluated similar to other randomized clinical trials; however, there are some questions that are specific for assessing the ACT component. These questions are best understood using the ASTIN trial as an example.
The specific methodologies used to make adaptive changes in the trial are important. In the ASTIN trial, allocation of treatment was provided by a central location at baseline, days 7, 21, and 90, using a computer generated Bayesian design algorithm. This algorithm determined the current real-time optimal dose and provided information regarding the need to vary the number of patients required, distribute the patients differently between placebo and the study drug, and correct the optimal dose of the study drug. An automated fax system (e-mail in today’s world) from other patients on the study drug provided information that was used to adjust and optimize the dose. In addition, an IDMC was established to utilize a termination rule for recommending discontinuation of the study after futility or efficacy was established. Termination for futility could be recommended only after a minimum of 500 evaluable patients had completed the study. A minimum of 250 evaluable patients were required before a recommendation could be made to terminate the study for efficacy reasons. Those patients with confirmed ischemic stroke by computerized axial tomography (CAT) scan who were still alive at study day 90 were termed evaluable patients.
Existing logistical issues and how they are being handled is important to know when evaluating an ACT study. In the ASTIN study, patient response information was transferred to a central location using an automated fax system. The adjusted dosing information was then sent back to the study site using this same fax system.
In ACTs, all proposed adaptive changes should be based on evidence and good clinical judgment. In the ASTIN study, the proposed adaptive changes were based on the dose-response information from other patients active and/or completed in the trial. The computer-generated specific adaptations that determined the current real-time optimal dose were provided by the Bayesian design algorithm. As mentioned earlier, information regarding the need to vary the number of patients required, distribute the patients differently between placebo and the study drug, and correct the optimal dose of the study drug was provided by this algorithm. A termination rule was established with specific criteria to be applied to the actual patient response information. This was developed to assist the IDMC in making a recommendation to discontinue the study after futility or efficacy was established.
It is important to know if extensive adaptation to the protocol occurred during the ACT.
When adaptation is extensive in efficacy confirming trials, the key hypothesis can become unclear and protection of the study’s integrity is at risk. The ASTIN study was a dose-response trial, not an efficacy-confirming trial. Adaptations of the dosing strength did occur and these were associated primarily with lack of efficacy or safety issues. The study’s integrity was not jeopardized.
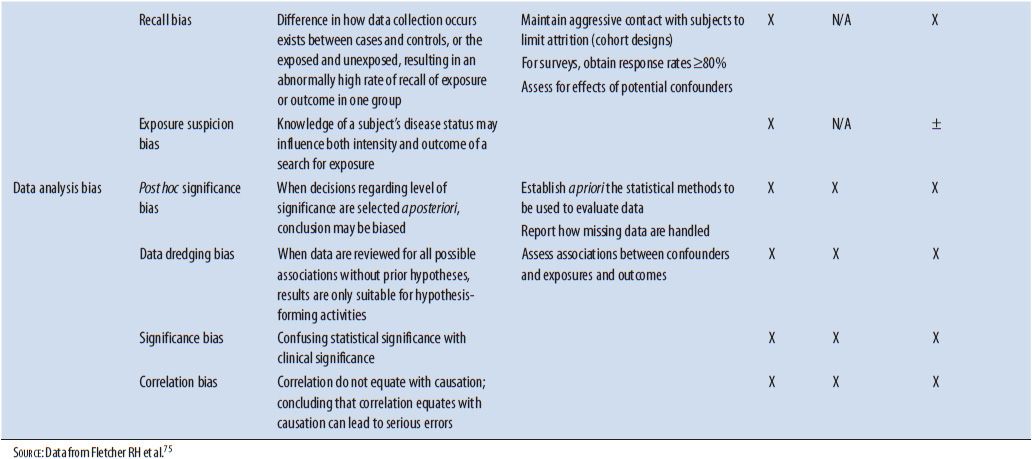
As with other study designs, obvious indications of bias entering the study and having effects on the results should be identified. Bias is a term used to describe a preference toward a particular result when this preference interferes with the ability to be impartial or objective. Several types of bias have been identified that can have major effects on the results of a study. See Table 5–3 for a list of biases from different causes that should be ruled out as having significant effects on the results of an ACT. Possible ways that bias could have entered the study should be identified and confirm the effect of that bias on the results determined. In the ASTIN study, several things were instituted to minimize the chance of bias. An IDMC consisting of three stroke clinicians and a statistician was established prospectively. This group worked independent of the study investigators and would make recommendations based on the patient information to the steering committee. For instance, changing doses based on patient safety and determining if the trial should be terminated according to predetermined criteria for efficacy or futility are two types of recommendations made by this committee. The executive steering committee consisting of expert stroke physicians also worked independent of the study investigators and monitored the conduct of the study, reviewed center performance, and made study decisions based on information provided by the IDMC. In addition, the study was double-blinded so neither the patient or study investigators were aware of what treatment the patient was receiving.
TABLE 5–3. TYPES OF BIAS
If an interim analysis was conducted, exactly who had access to the information created from the interim analysis and how could this have affected any of the results are crucial to know. As mentioned previously, the ASTIN study had an IDMC formed to conduct interim analyses on the patient information received from the ongoing study. Based on the description of this group in the article, it would appear members of that committee had few, if any, interactions with Pfizer, the company sponsoring the trial.
Sometimes the ACT is stopped early based on an interim analysis. A determination should be made if stopping the trial early had any effect on the results that would prevent development of strong conclusions. If the discontinuation significantly shortens the period of treatment, evidence may be lacking for any conclusions to be made. The ASTIN study was stopped early due to lack of the test drug’s dose response in efficacy. The effect on the evidence was minimal since specific numbers of patients needed to be enrolled before discontinuation could even be considered were predetermined before the study was initiated. A total of 551 evaluable patients had been enrolled (stopping rule required a minimum of 500 evaluable patients), received some dose of the study drug within the allotted range, and completed the study to day 90 before the trial was discontinued. An adequate sample size appeared to be obtained to meet the set power.
The benefits of ACT design are obvious and this encourages further exploration for appropriate application to clinical programs. Adaptations to number of patients, eligibility criteria, drug dose, and randomization allocation can significantly improve the efficiency of drug development and overall patient care. This is an area of study design research that is being implemented more frequently for specific uses. See Chapter 8 for further discussion of ACT.
Appendix 5–1 contains a list of additional questions specific to ACTs. These specific questions are in addition to the standard questions used to evaluate other randomized clinical trials noted in Chapter 4 and using basic tools like the Ten Major Considerations Checklist discussed in the Introduction section.
STABILITY STUDIES/IN VITRO STUDIES
Stability studies determine the stability of drugs in various preparations (e.g., ophthalmologic, intravenous, topical, and oral) under various conditions (e.g., heat, freezing, refrigeration, and room temperature). Stability of a pharmaceutical product is defined by the United States Pharmacopeia (USP) as “extent to which a product retains, within specific limits and throughout its period of storage and use (i.e., its shelf-life), the same properties and characteristics that it possessed at the time of its manufacture.”49 These studies are extremely important to the practice of pharmacy. For example, pharmacists who prepare intravenous solutions for use by patients at home often want to know how long a drug admixed in a particular solution is stable. Another stability question could be whether the length of time to maintain stability would be increased with freezing the admixture. This information helps determine how many intravenous admixtures may be dispensed at a time. It is also important for pharmacists involved with extemporaneous compounding to know the length of time a particular preparation is stable.
Stability study requirements and expiration dating are presented in the Current Good Manufacturing Practices (cGMPs),50 the USP,51 and the FDA and International Conference on Harmonization (ICH) guidelines. The listings for FDA Guidelines associated with various stability testing can be located at http://www.fda.gov/drugs/guidancecomplianceregulatoryinformation/default.htm. The ICH guideline Web page is located at http://www.ich.org/products/guidelines/quality/article/quality-guidelines.html. The cGMPs require a written testing program designed to assess the stability characteristics of drug products.50 The USP contains detailed stability and expiration dating information in addition to stability considerations in dispensing practices.51 Design of stability studies to acquire expiry and product storage, in addition to directions for submission of this information, is found in the FDA guidance documents. The ICH guidelines provide common approaches used by the pharmaceutical industry for assessing stability of drug products.
Generally, stability studies are not published but remain unpublished data on file with the pharmaceutical company or compounding pharmacy and in some cases not available to the public since they represent trade secrets. Trissel and associates have provided study guidelines for assessing the quality of stability studies that do get published.52 These guidelines state that investigators conducting stability studies should provide:
•
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


