Liquids, liquid crystals, and ionic liquids
4.1 Liquid formation
Decreasing the temperature or increasing the pressure on a gas will lead to condensation. Most commonly, a gas condenses directly to the liquid phase. At one atmosphere of pressure, the temperature of this phase transition is called the normal boiling point, while at a pressure of 1 bar (the standard state pressure p°) this temperature is called the standard boiling point. Decrease the temperature yet further at the standard pressure, and the liquid at equilibrium would crystallize into a solid at the standard freezing point Tf (also called the standard melting temperature or standard melting point). Because of the very small change in molar volume between the liquid and solid phases, the normal and standard freezing points are essentially equal.
4.2 Properties of liquids
The liquid phase is a fluid, just as a gas is, and will take on the shape of its container. However, it is a much different type of fluid because of intermolecular interactions and the close proximity of the molecules within it. This means that liquids are almost incompressible and that many of their properties can be thought of in terms of a continuous material. That is, as long as we do not look on too fine a scale, many properties of a liquid can be approximated in terms of a continuous distribution of matter rather than discreet molecules.
The density of a liquid is much greater than that of a gas. The molar volume of an ideal gas at standard ambient temperature and pressure (SATP, T = 298.15 K, p = 1 bar = 100 kPa) is

The molar volume of liquid water at SATP is

Therefore, the ratio of the volume of water vapor (acting ideally) to water at SATP is 1370. This is a general observation: the molar volume decreases by a factor of about 1000 upon condensation. Therefore, the mean distance between molecules changes by roughly a factor of 10.
Dividing these values of molar volume by NA, we find the mean volume occupied by a molecule. The inverse of this quantity is the number density C, which is found in the last column of Table 4.1. Assuming the molecules are spherical with radius r, their volume is  . Thus, a rough estimate for the radius of the sphere ‘occupied’ by a water molecule is 21.4 Å in the gas phase and 1.93 Å in the liquid phase. Obviously, a molecule does not shrink by a factor of 10 upon condensation. The molecules are the same size in both cases, but the mean distance between them changes.
. Thus, a rough estimate for the radius of the sphere ‘occupied’ by a water molecule is 21.4 Å in the gas phase and 1.93 Å in the liquid phase. Obviously, a molecule does not shrink by a factor of 10 upon condensation. The molecules are the same size in both cases, but the mean distance between them changes.
Table 4.1 Characteristics of various liquids: molar mass M, melting point Tf, boiling point Tb, mass density ρ, molar volume Vm, number density
C. Values for ρ and Vm refer to 298.15 K except for Ar (90 K), CH4 (90.68 K), H2 (20.00 K), HF (293.15 K), He (4.30 K), N2 (78 K), NH3 (239.15 K), O2 (90.00 K), NaCl (1074 K), Ag (1235 K), Na (371 K), and Re (3453 K). Values taken from Poling, Prausnitz and O’Connell (see Further Reading) or the 96th edition of the CRC Handbook of Chemistry and Physics.
| Species | M / g mol−1 | Tf / K | Tb / K | ρ / kg m−3 | Vm × 106 / m3 mol−1 | C × 10−28 / m−3 |
|---|---|---|---|---|---|---|
| Ar | 39.948 | 83.80 | 87.27 | 1373 | 29.10 | 2.070 |
| Br2 | 159.808 | 265.85 | 331.90 | 3102 | 51.52 | 1.169 |
| CH4 | 16.043 | 90.69 | 111.66 | 451.4 | 35.54 | 1.694 |
| C2H5OH | 46.069 | 159.05 | 351.80 | 785.1 | 58.68 | 1.026 |
| C6H6 | 78.114 | 278.68 | 353.24 | 873.7 | 89.41 | 0.674 |
| H2 | 2.016 | 13.56 | 20.38 | 71.01 | 28.39 | 2.121 |
| HF | 20.006 | 189.58 | 292.68 | 966.9 | 20.69 | 2.911 |
| H2O | 18.015 | 273.15 | 373.15 | 997 | 18.07 | 3.333 |
| He | 4.003 | 2.15 | 4.30 | 123 | 32.54 | 1.850 |
| NH3 | 17.031 | 195.41 | 239.82 | 682.3 | 24.96 | 2.413 |
| N2 | 28.014 | 63.15 | 77.35 | 804.1 | 34.84 | 1.729 |
| O2 | 31.999 | 54.36 | 90.17 | 1149 | 27.85 | 2.162 |
| NaCl | 58.44 | 1074 | 1738 | 1556 | 37.56 | 1.603 |
| Ag | 107.868 | 1235.08 | 2485 | 9320 | 11.57 | 5.203 |
| Na | 22.9898 | 370.96 | 1156.1 | 927 | 24.80 | 2.428 |
| Hg | 200.59 | 234.28 | 629.73 | 13534 | 14.82 | 4.063 |
| Re | 186.2 | 3453 | 5900 | 18900 | 9.85 | 6.113 |
We know that, compared to gases, liquids are nearly incompressible. Their volume – and therefore their density – changes slowly as temperature changes in comparison to the large changes observed for a gas. In an ideal gas the relationship is

An inverse relationship between density and temperature is generally observed for liquids; however, the functional form – known as the Tait equation – is rather complex. For a limited range above the melting point, the density of liquid metals and molten salts follows the equation

where ρm is the density of the liquid at the melting point, k is a constant and Tm is the maximum temperature for which the linear relationship holds. The value of k usually falls in the range of 0.5 to 8 kg m−3 K−1.
Water is a very unusual liquid but also the most important. Its maximum density of 999.9749 kg m−3 occurs at 277.15 K, that is, 4 K above Tf. Liquid water, like many liquids, can also be supercooled (cooled below Tf ) if special precautions (slow cooling, purity, absence of particulates) are taken. An empirical fit to the mass density of water over the entire measured temperature range shown in Fig. 4.1 requires a fourth-order polynomial,

where k0 = −4240, k1 = 63.3, k2 = −0.286, k3 = 5.73 × 10−4, and k4 = −4.34 × 10−7.

Figure 4.1 The mass density and molar volume of normal water and supercooled water as a function of temperature. Values taken from the 96th edition of the CRC Handbook of Chemistry and Physics.
4.3 Intermolecular interaction in liquids
As shown in Table 4.1, which pertains to standard pressure, the liquid phase is stable over much different temperature ranges depending on the nature of the intermolecular forces in the material under consideration. The intermolecular interactions discussed previously that lead to non-ideality in gases are more strongly present in liquids because the molecules are closer together. The interactions depend on the types of molecules present. The interactions in liquids of inert gas atoms or nonpolar molecules are dominated by pairwise-additive terms. Many-body terms are negligible. Polar liquids usually have substantial many-body terms while liquid metals possess very large many-body terms. Constructing a potential energy function to describe many-body interactions is much more complicated than constructing one with just pairwise-additive terms.
The intermolecular interactions of a liquid are perhaps the most difficult to treat of the three common phases. In a gas, the distance between molecules allows us to ignore interactions in the ideal state and then add interactions as a small perturbation to the ideal model. In the solid state, the perfect order of a crystal introduces symmetry, which greatly simplifies the treatment of interactions. The result of these difficulties is that fundamental theories of the properties of liquids have been the most difficult to advance and lag considerably behind those of the gas and solid phases.
Liquid metals and He are exceptional liquids. Helium has two liquid phases, which is also observed for P but still debated for H2O and S. Helium is, however, the only elemental liquid that cannot be solidified at 1 atm pressure (25 atm are required for solidification), which means it has no liquid/solid phase boundary at atmospheric pressure. Liquid metals are electrically conductive, which indicates that electrons are delocalized because of metallic bonding. Delocalization is a manifestation of the many-body interactions mentioned above. Most molten salts are composed of ions, but there are exceptions such as HgCl2, which has low electrical conductivity and, thus, must be composed of uncharged species.
H2O is the only naturally occurring inorganic liquid on Earth. It is a truly exceptional liquid. Indeed, if it were not the most important liquid, we might not even study its properties because of the complications of dealing with a heavily hydrogen-bonded liquid with an unusually large relative permittivity that readily autoionizes. However, as it is the most important liquid we will consequently spend considerable effort trying to understand it, and solutions in which it is a solvent.
Our first-order model of a liquid begins with assuming that the molecules or atoms which comprise the liquid are nondeformable spheres with a fixed diameter. Their interactions with other molecules are independent of orientation. This is known as a hard-sphere model. A hard-sphere potential gives a good approximation to the thermal conductivity and viscosity of a fluid. This is because these properties are mainly determined by long-range repulsions rather than van der Waals attractions. Condensation of a gas into a liquid requires a potential with an attractive well. A simple and widely used semi-empirical potential is the Lennard–Jones potential (also known as L-J or 6-12 potential). It is defined in terms of the interparticle separation R and has the form

where σ is an empirically determined hard-sphere diameter of the atom/molecule, ϵ is related to the well depth (minimum of the potential), m = 12, and n = 6. Examples of Lennard–Jones potentials for the noble gases are shown in Fig. 4.2. The shape should be familiar, as they look much like the attractive potential that is associated with the formation of a chemical bond. The potentials approach zero asymptotically as they approach infinite separation. They pass through a minimum and then again equal zero when the separation is equal to the hard-sphere diameter, that is, at R = σ. They approach infinity as the intermolecular separation decreases below the effective molecular diameter because two molecules cannot occupy the same space.

Figure 4.2 The Lennard–Jones potentials for He, Ne, Ar, Kr, and Xe.
The Lennard–Jones parameters for a number of gases are listed in Table 4.2, where some expected trends are immediately obvious. In going from He down the column to Xe, the size of the atom increases, as does its polarizability. Thus, the hard-sphere diameter σ increases as does the well depth ϵ. Similar trends occur for the hydrogen halides and halogens, except for the well depths of HI and I2, which are unusually small. The substitution of F for H in either methane or silane leads to a larger molecule but also one with a smaller well depth. Changing from a triple bond to a double bond to a single bond in the C2H2−C2H4−C2H6 series leads to a larger molecule, but also to one that is successively less polarizable and more weakly interacting. Benzene is more compact than cyclohexane, but its extended system of π electrons leads to stronger intermolecular interactions. The difference between benzene and cyclohexane is much larger than the difference between ethene and ethane. Methanol is smaller and much more polar than ethanol; such greater polarity leads to a much greater well depth for methanol. Substituting a polar OH group for a nonpolar H makes the intermolecular interactions of C2H5OH much stronger than those of C2H6.
Table 4.2 Parameters for Lennard–Jones potentials determined from viscosity data. Values from Poling, Prausnitz and O’Connell.
| Formula | Molecule | σ / Å | ϵ/kB / K |
|---|---|---|---|
| He | helium | 2.551 | 10.22 |
| Ne | neon | 2.820 | 32.8 |
| Ar | argon | 3.542 | 93.3 |
| Kr | krypton | 3.655 | 178.9 |
| Xe | xenon | 4.047 | 231.0 |
| H2 | hydrogen | 2.827 | 59.7 |
| H2O | water | 2.641 | 809.1 |
| HF | hydrogen fluoride | 3.148 | 330 |
| HCl | hydrogen chloride | 3.339 | 344.7 |
| HBr | hydrogen bromide | 3.353 | 449 |
| HI | hydrogen iodide | 4.211 | 288.7 |
| HCN | hydrogen cyanide | 3.630 | 569.1 |
| F2 | fluorine | 3.357 | 112.6 |
| Cl2 | chlorine | 4.217 | 316.0 |
| Br2 | bromine | 4.296 | 507.9 |
| I2 | iodine | 5.160 | 474.2 |
| NH3 | ammonia | 2.900 | 558.3 |
| N2 | nitrogen | 3.798 | 71.4 |
| NO | nitric oxide | 3.492 | 116.7 |
| NO2 | nitrous oxide | 3.828 | 232.4 |
| O2 | oxygen | 3.467 | 106.7 |
| SF6 | sulfur hexafluoride | 5.128 | 222.1 |
| SiH4 | silane | 4.084 | 207.6 |
| SiF4 | tetrafluorosilane | 4.880 | 171.9 |
| UF6 | uranium hexafluoride | 5.967 | 236.8 |
| CH4 | methane | 3.758 | 148.6 |
| CF4 | tetrafluoromethane | 4.662 | 134.0 |
| CCl4 | tetrachloromethane | 5.947 | 322.7 |
| C2H2 | ethyne | 4.033 | 231.8 |
| C2H4 | ethene | 4.163 | 224.7 |
| C2H6 | ethane | 4.443 | 215.7 |
| C6H6 | benzene | 5.349 | 412.3 |
| C6H12 | cyclohexane | 6.182 | 297.1 |
| n-C6H14 | n-hexane | 5.949 | 399.3 |
| CH3OH | methanol | 3.626 | 481.8 |
| C2H5OH | ethanol | 4.530 | 362.6 |
| CO | carbon monoxide | 3.690 | 91.7 |
| CO2 | carbon dioxide | 3.941 | 195.2 |
For any combination of m and n for which m > n, the depth of the minimum is
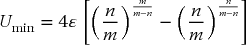
and it occurs at

The R−6 term accurately represents the long-range limit of the dispersion energy. The R−12 term is a convenient-for-integration term that approximates the short-range repulsion. Because the exchange energy actually is better approximated by an exponential function, a more accurate representation is the (exp-6) potential

As a first step to understanding the structure of a liquid, let us calculate the potential energy of hard spheres interacting only through a potential that can be described by a Lennard–Jones potential, as shown in Fig. 4.2. Consider N molecules described by the potential U(R). The molecules only interact through pairwise-additive terms, where R is the distance between their centers. The potential energy of their interaction vanishes at R = σ, which defines the effective diameter of the molecules. The molecules are labeled 1 to N, and we consider two of them, i and j. The number of distinct pairs ij is  . U(Rij) is the potential energy of the interaction between pair ij. Since we have assumed that the molecules in the liquid only interact in a pairwise fashion, in order to calculate the total potential energy of the liquid, Φ, we need to sum the contributions of all the pairs, being careful not to double-count any pairs (indicated by the i > j in the summation),
. U(Rij) is the potential energy of the interaction between pair ij. Since we have assumed that the molecules in the liquid only interact in a pairwise fashion, in order to calculate the total potential energy of the liquid, Φ, we need to sum the contributions of all the pairs, being careful not to double-count any pairs (indicated by the i > j in the summation),

Now we try to build upon this simple starting point to understand the structure and properties of the liquid phase.
4.3.1 Directed practice
Explain the trend in the Lennard–Jones parameters of CH4, CF4, and CCl4.
4.4 Structure of liquids
The structure of a liquid can be approached from two extreme limits. One is to think of it as an extremely dense gas, while the other is to start from the vantage of a disordered solid. To the first model, we must add intermolecular interactions and the potential for many-body interactions. To the second we need to add transport and allowance for short-range order but the lack of long-range order.
To describe the structure of a liquid we introduce two parameters. One is the coordination number, Z; this is the mean number of nearest neighbors. The second is the radial distribution function g(R), also known as the pair correlation function.
In a fluid with no intermolecular forces, every molecule moves through the entire volume of the sample, independent of the presence of any other molecule. The fluid would be homogeneous. For a given volume element ΔV, there is an equal probability of finding a molecule in this volume element anywhere in the total volume V. The probability of finding a molecule in the volume element is ΔV/V. The probability of finding one particular molecule in the volume ΔV at point R1, and another particular molecule in the volume ΔV at point R2, is then the product (ΔV/V)2 independent of R1 and R2. If there are N total molecules, then the probability of simultaneously finding any molecule in the volume element at R1 and any other molecule in ΔV at R2 is N(N − 1)(ΔV/V)2. Again, this is independent of R1 and R2. Thus, the joint probability density of finding any molecule at R1 and any other molecule at R2 is

where n is the conditional probability density of finding a molecule a distance R from any arbitrarily chosen molecule. For the large number of molecules in a macroscopic sample,n = N/V = C0, where C0 is the mean number density averaged over the entire liquid. This probability density is independent of R. We conclude that, in the absence of intermolecular interactions, the fluid is structureless. That is, the probability of finding a pair of molecules with any arbitrarily chosen separation R is independent of R. In a structureless fluid the radial distribution function is constant, g(R) = 1.
Consider a liquid composed of spherical molecules with a diameter σ. The radial distribution function describes how many molecules are at a given center-to-center distance R from the reference molecule. Because the molecules have a finite size, there is now a strong short-range repulsive interaction between them that excludes one molecule from the volume occupied by the other. As can be seen in Fig. 4.3, this repulsion has a huge influence on the structure of the liquid. The radial distribution function must start at zero, after which at separations greater than σ it increases. Several maxima are observed, with the first occurring near the minimum in U(R) – that is, at the first nearest-neighbor distance. In a perfect lattice of spheres of the same diameter, the nearest neighbors are all equidistant from the reference atom. The next nearest neighbors form a second shell and reside at a larger distance, and so on. The radial distribution function records these positions as peaks in a plot of g(R) versus R. Long-range order in a solid is indicated by an infinite series of such peaks, and thermal motion broadens the peaks. The introduction of disorder turns the series of peaks into a continuous distribution. If the sample is amorphous, as in a liquid, then there is short-range order but a lack of long-range order. This is reflected in g(R) by a decay in the height of the broad peaks, as shown in Fig. 4.3. Usually, at distances greater than roughly three molecular diameters, the oscillations disappear and g(R) converges on 1.

Figure 4.3 A prototypical radial distribution function for a disordered liquid as compared to a homogeneous liquid.
Let us now define the radial distribution function mathematically, which will allow us to develop a rigid sphere model of the liquid. This model was refined by Kirkwood in 1935. We place a molecule at the origin O and measure the radial distance R from this origin. We define the number density C(R) as the number of molecules per unit volume at distance R. The radial distribution function is the ratio of the number density as a function of R to the mean number density C0,

As can be seen in Fig. 4.3, g(R) exhibits damped oscillations that vary from slightly above the mean density to below it outside of the diameter of the molecule at the origin. The radial distribution function is determined by X-ray or neutron diffraction data. Its value represents a time-averaged quantity. At any given distance over a period of time there will be significant fluctuations in g(R) because of the constant thermal motion of the molecules.
This constant thermal motion means that while the time-averaged distance between one molecule and its nearest neighbor is constant, the identity of the two molecules is not. Recall that each molecule in the liquid is able to sample the entire volume of the liquid. If we were to momentarily stop the molecules and take a picture, any two pictures would look almost identical. But if we were to take a picture, label the molecules, then follow their motions as a function of time, we would see that the configuration of molecules changes on an extremely rapid time scale. The molecule initially at the origin will move and be replaced by another, and the molecules that initially formed the set of nearest neighbors exchange their places with other molecules. The time scale for this decay of the initial structure is several picoseconds (10−12 s).
The study of g(R) as a function of temperature and pressure reveals much about the nature of a liquid. The structure of a liquid does not depend strongly on temperature; rather, it is determined mainly by the density. We can now define the coordination number Z in terms of the radial distribution function

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


